











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
1. Introducción
La investigación en el desarrollo de nuevas moléculas que optimicen una propiedad deseada se ha vuelto una necesidad en casi todos los sectores productivos como el alimenticio, el agropecuario, la salud y la industria, debido a que estos buscan aumentar la efectividad de dichas moléculas.
Los esfuerzos de la química sintética a lo largo del ultimo siglo ha logrado producir más de 60 millones de compuestos [1] y el número de moléculas pequeñas, candidatos a fármacos asciende a más de 100 millones de moléculas diferentes [2]. Este gran número de moléculas a explorar es llamado espacio químico. El número de posibilidades es inmenso, incluso cuando este espacio se limita solamente a moléculas basadas en el modelo de Lipinski [3]. Este modelo establece, que para que un compuesto pueda ser suministrado oralmente, debe cumplir con al menos tres de las siguientes condiciones: (a) poseer un peso molecular por debajo de 500; (b) no tener más de 5 átomos donores de enlace de hidrógeno; (c) poseer un coeficiente de partición octanol-agua debajo de 5; y (d) no tener más de 10 átomos aceptores de enlace de hidrógeno.
A pesar de esta gran cantidad de estructuras químicas, se ha vuelto cada vez más difícil desarrollar nuevos medicamentos, en gran parte debido a la falta de eficacia, losefectos secundarios y los problemas de toxicidad que se presentan en su desarrollo [4].
Independientemente de la gran cantidad de moléculas disponibles el ritmo de aprobación de nuevos fármacos ha disminuido notablemente, dejando espacios para que nuevos métodos o técnicas que pueden mejorar el proceso actual [5]. Por ejemplo, las tecnologías in silico permiten acelerar el descubrimiento de nuevos fármacos y reducir los gastos que conlleva el trabajo experimental. Esta reducción de gastos se logra mediante la química computacional y mediante la creación de programas de predicciones con la ayuda de la bioinformática y la bioestadística [6].
Un método moderno para descubrir nuevas moléculas es el aprendizaje automático 7 junto con él las redes neuronales artificiales 8. Un tipo particular de red neuronal artificial y muy utilizada para resolver múltiples problemas prácticos en los que se requiere procesar una gran cantidad de imágenes o señales de gran tamaño son las redes neuronales convolucionales (RNC) [9-11].
En este trabajo, se analizó el aprendizaje del uso terapéutico de fármacos a partir de su configuración estereoquímica usando redes neuronales convolucionales. Se escogieron 12 usos terapéuticos: Antiinfeccioso, Antiinflamatorio, Antineoplásico, Cardiovascular, Sistema nerviosos central (snc), Dermatológico, Gastrointestinal, Hematológicos, Regulación de lípidos, Control reproductivo, Sistema respiratorio, Urológicos [12].
La capacidad de los modelos de aprendizaje automático (in silico) para predecir la acción de las moléculas, ayudaría a reducir considerablemente el número de moléculas a probar empíricamente, ya que priorizaría las que el modelo prediga. De esta manera, se aumentaría la velocidad en la búsqueda del medicamento más eficaz y disminuiría los costos atribuidos a las pruebas empíricas.
En este trabajo se aborda el problema del aprendizaje de funciones de fármacos a partir de estructuras químicas. Recientemente se han evaluado dos métodos de clasificación de fármacos derivados de la estructura química: imágenes químicas con redes neuronales convolucionales [13] y huellas dactilares [14] moleculares con bosques aleatorios [15]. Los resultados que usaron la imagen química superaron las predicciones que utilizaron cambios transcriptómicos inducidos por fármacos como representaciones químicas (huellas dactilares). Ellos sugieren que la imagen de la estructura de una sustancia química tal como se muestra en la Fig. 1 contiene al menos tanta información sobre su uso terapéutico como la respuesta celular transcripcional a esa sustancia química [16].

Fuente: National Library of Medicine (NLM).
Figura 1 Imagen 2D de la estructura química de la Aspirina.
Dentro de los enfoques usados en la comunidad del Docking [17] hay uno en particular que usa la técnica de parejas, el cual describe la proteína y el ligando [18] como superficies complementarias [19,20].
Esta complementariedad geométrica es un método que describe la proteína y el ligando como un conjunto de rasgos que los hace acoplables [21]. Estos rasgos pueden incluir la superficie molecular, o descriptores de la complementariedad de la superficie. En este caso, la superficie del receptor molecular es descrita en términos de su área superficial de accesibilidad solvente y la del ligando es descrita en sus términos de descripción a su ajuste a la superficie.
Basados en este enfoque de complementariedad de forma, en este trabajo nosotros proponemos considerar la información de la forma tridimensional de la estructura molecular, para el predecir la función de los fármacos [22]. Esta idea se sustenta en el hecho de que el ligando y proteína tienen forma tridimensional y el acople del ligando a una proteína en particular debería depender de su forma tridimensional.
Es así, que para predecir la función de un fármaco se propone en este trabajo entrenar una red neuronal convolucional con diferentes vistas de su estructura molecular tridimensional, tal como se puede observar en la Fig. 2.

En este trabajo, utilizamos el aprendizaje automático y las representaciones de las moléculas químicas en 2D y 3D de 12 categorías de uso terapéutico derivadas de MeSH [23] (en inglés, MeSH, Medical Subject Headings) para predecir y clasificar las clases de usos terapéuticos como se observa en la Fig. 3.

2. Metodología
Para la construcción de un sistema de reconocimiento para predecir la función de los fármacos de uso terapéutico a partir de su estructura química se puede optar por el uso de técnicas de extracción de las características manuales para luego alimentar un modelo de aprendizaje automático. En este trabajo se propone el uso de redes neuronales convolucionales, dado que permiten la extracción de las características de las estructuras química de forma automática.
Investigaciones realizadas en la misma área por Aliper et al. [13], Gitter et al [24] y Ragoza en al [25] proponen una labor de ensayo y error para encontrar los hiperparámetros adecuados de la red que permiten su buen entrenamiento, partiendo de algunos valores que han dado buen resultado en trabajos similares. Es por ello, que este trabajo se enmarca en el tipo de investigación experimental.
En este trabajo se realizó el estudio con una red neuronal convolucional para lograr predecir la función de fármacos de uso terapéutico, utilizando para su entrenamiento la información tridimensional de su estructura molecular.
Este trabajo fue inspirado en el artículo de Antony Gitter et al, los cuales usaron imágenes 2D de la estructura molecular de fármacos para 12 usos terapéuticos. Nosotros hemos escogido también los mismos 12 usos terapéuticos.
Los fármacos para las 12 categorías de uso terapéutico fueron seleccionados de la Biblioteca Nacional de Medicina y Centro Nacional de Información Biotecnológica, ubicados en la pagina web PubChem [26] siguiendo la siguiente ruta: categoría productos químicos y drogas - acciones farmacológicas - usos terapéuticos.
En esta sección describiremos como fue construida la base de datos con la información 2D de la estructura molecular y cómo fue construida la base de datos con la información 3D.
2.1. Construcción de la base de datos con imágenes 2D de la estructura molecular del fármaco
Para generar las imágenes 2D de la estructura molecular de los fármacos, se realizó el procedimiento sugerido por Antony Gitter et al:
Los identificadores CIDs de cada molécula se convirtieron a cadenas SMILES [27] usando el paquete de Python pubchempy (https://github. com/mcs07/PubChemPy).
Se excluyeron sustancias químicas de múltiples clases, para permitir una comparación directa.
La lista final se filtró para eliminar múltiples versiones de moléculas que difieren sólo por las sales que las acompañan.
Cada una de las imágenes 2D obtenidas tienen tres canales (RGB) con un tamaño de 500 x 500. En la Fig. 4 se muestra un ejemplo de la imagen 2D obtenida para la molécula de la Aspirina. En la Fig. 5 se observa mejor la frecuencia de fármacos por cada clase

Fuente: National Library of Medicine (NLM).
Figura 4 Imagen 2D de la estructura química de la Aspirina.

Por otro lado, se observa en la gráfica 5 que la base de datos obtenida se encuentra muy desbalanceada. Esto se debe a que en los usos terapéuticos escogidos de la base datos de PubChem existen más fármacos en una clase que en otra.
2.2. Aumentación digital de la base de datos con imágenes 2D de la estructura molecular del fármaco
Para aumentar la base de datos de imágenes 2D se crearon más imágenes a partir de transformaciones digitales. Para realizar esta aumentación se utilizó el paquete Augmentor [28] de Python [29] con el cual se crearon nuevas imágenes a partir de las imágenes originales que contiene cada clase.
Para hacer el sistema de predicción invariante a la rotación y a la escala de la imagen 2D de la molécula, nosotros en este trabajo hemos escogido realizar solamente dos transformaciones: rotación y acercamiento.
Las imágenes a rotar se escogieron de forma aleatoria con las siguientes condiciones:
Rotación: Las imágenes fueron rotadas entre 10 y 15 grados con respecto al originen, con una probabilidad de realizar la rotación de 0.7.
Acercamiento: A las imágenes que se le aplicó esta transformación tuvieron un acercamiento del 0.01% y 0.03%, con una probabilidad de realizar el acercamiento de 0.5.
Para algunas imágenes el comando le aplico ambas transformaciones.
La Fig. 6 muestra un ejemplo de la rotación realizada a una de las moléculas.

Fuente: Elaborado por los autores.
Figura 6 Ejemplo de rotación realizada a una imagen a) original b) rotación de 15º
La Fig. 7 muestra la frecuencia de las imágenes 2D obtenidas por cada clase después de realizada la aumentación digital. En total se crearon 4235 imágenes nuevas para un total de 10717 imágenes 2D.

1.1 Construcción de la base de datos con imágenes 3D de la estructura molecular de los fármacos
Para construir una base de datos que aporte información tridimensional de la estructura molecular, la representación 3D de la estructura molecular que hemos escogido es Ball and Stick [30] porque las barras son más visibles para representar el tipo de enlace y las bolas y su respectivo color diferencian mejor el tipo de átomo.
Para construir tal base de datos nosotros hemos decidido generar imágenes 2D con diferentes vistas de la imagen 3D de la molécula. Para escoger el numero de vistas a realizar para cada una de las moléculas nos hemos planteado la siguiente pregunta: ¿cuántas vistas son necesarias para reconstruir la estructura molecular 3D de una molécula? La respuesta no es única porque depende de la complejidad de la estructura 3D de la molécula. Nosotros en este trabajo hemos escogido solamente las 6 vistas que se obtienen al realizar las proyecciones sobre cada una de las caras del paralelepípedo en donde se inscribe la molécula. La Fig. 8 muestra un ejemplo del paralelepípedo donde se inscribe una molécula. Hubiésemos deseado construir más vistas para obtener más información del fármaco y seguramente un mejor rendimiento de la RNC; pero nos vimos limitados por nuestra capacidad actual de computo. Sin embargo, con el número de vistas tomado se pudieron contestar las preguntas de investigación.

Fuente: Elaborado por los autores.
Figura 8 Ejemplo de imagen 3D de la estructura molecular del Ácido acetilsalicílico.
El proceso para la creación de la base de datos con las seis vistas en 3D de las moléculas siguió los siguientes pasos:
El primer paso fue organizar la base de datos de cada una de las clases en un archivo de Excel por fila con la estructura 2D de cada una de las moléculas descritas con su cadena de SMILES.
El segundo paso fue realizar la transformación de las estructuras 2D a 3D. Para este proceso se utilizó una conexión online del software CORINA del Intituto Nacional de Cancer (NIH: National Cancer Institute). Cada uno de los SMILES se transformó a un archivo de dato SDF (Spatial Data File).
El tercer paso consistió en la creación de las 6 vistas. Para ello se utilizó el software MATLAB versión R2018a (9.4.0.813654) para leer los archivos SDF y realizar una visualización 3D. Luego, se realizaron rotaciones sobre el eje x y y en cada rotación se guardo la imagen 2D correspondiente. Sobre el eje x se realizó una rotación cada 90º hasta completar 360º y sobre eje y primero se hizo una rotación de 90º y luego de 180º.
Un ejemplo de las vistas 2D obtenidas para la molécula de la Aspirina se muestra en la Fig. 2. Este proceso se realizó de forma individual para los 6956 archivos SDF generados con CORINA. Algunos archivos SDF no fueron admitidos por el software CORINA, por razones desconocidas, por lo tanto, el número total de imágenes generadas fue de 21397.
La Fig. 9 muestra la frecuencia de las imágenes 3D obtenidas por cada clase después de realizar las seis vistas. En total se crearon 21397 vistas 2D con la imagen tridimensional de cada molécula.

En la Tabla 1 se muestra la distribución de las imágenes 2D y 3D de las moléculas por cada una de las clases de fármacos.

Primero presentaremos los resultados obtenidos con una RNC desde cero. Usando inicialmente la base de datos con imágenes 2D y luego usando la base de datos con imágenes 3D.
Luego, presentaremos los resultados usando transferencia de aprendizaje. inicialmente usamos la base de datos con imágenes 2D, luego usando la base de datos 2D aumentada digital y finalmente con la base de datos con imágenes 3D.
Finalmente, presentamos los resultados obtenidos en el pronóstico del uso terapéutico de un fármaco al cual se le ha comprobado que tiene dos principios activos.
La métrica utilizada para comparar cada uno de los modelos fue la exactitud ya que nos interesa saber que porcentaje de fármacos clasifica correctamente; y para validar el rendimiento del modelo para cada una de las clases de uso terapéutico se utilizó la precisión ya que nos indica que cuando los clasifica positivos que porcentaje clasifica correctamente.
3. Resultados
3.1. Redes neuronales desde cero
En el entrenamiento de esta red las imágenes fueron reescaladas a un tamaño de 250 x 250, las funciones de activación usadas fueron ReLu en las capas ocultas y Softmax como capa de salida, la función de pérdida utilizada fue la entropía cruzada, el optimizador utilizado fue el Adamax con una tasa de aprendizaje de 0.001.
3.2. Resultados con base de datos 2D
Con este modelo se logró una Exactitud del 52\%. Aunque el resultado no fue el mejor, el modelo logra predecir el uso terapéutico de algunos fármacos a partir de su representación 2D.
3.3. Resultados con base de datos 3D
Con este modelo se logró una Exactitud del 61\%. Aunque el resultado no fue el mejor, se muestra que hubo una mejoría en la predicción del uso terapéutico de algunos fármacos a partir de su representación 3D.
3.4. Resultados con transferencia de aprendizaje
Para la transferencia de aprendizaje se usaron los pesos de ResNet-50, una red neuronal convolucional con 50 capas, distribuidas en 32 capas de convolución normal, 16 capas de convolución separables en profundidad y dos capas de convolución densamente conectadas. Las imágenes fueron re-escaladas a un tamaño 224 x 224, usando ReLu como función de activación en las capas profundas y SoftMax en la capa de salida, el padding utilizado fue same para obtener la misma dimensión cuando se apliquen los filtros, para controlar el cambio de distribución se aplicó una capa de normalización, la cantidad de épocas programadas fue de 50 con un tamaño por lote de 16, en este entrenamiento no se aplicó aumentación de datos.
Esta arquitectura se usó tanto para la base de datos con imágenes 2D como para 3D.
3.5. Resultados con base de datos 2D sin aumentación de datos
Con la base de datos 2D el mejor resultado se logró con una tasa de aprendizaje de 0.001. La Fig. 10 muestra en la matriz de confusión obtenida.

Fuente: Elaborado por los autores.
Figura 10 Matriz de confusión con transferencia de aprendizaje (ResNet50) con imágenes 2D.
Se puede observar que este modelo no clasificó ningún fármaco ni para uso terapéutico del sistema respiratorio ni urológico. Este resultado puede deberse a que estas clases son las que tienen menos imágenes 2D de fármacos. Aunque la clase de los medicamentos gastrointestinales a pesar tener una menor cantidad de fármacos que los antiinflamatorios tuvo una mejor predicción. Por otro lado, la clase que mejor predijo fue los fármacos antiifecciosos con un 77%. Esta clase es la que más imágenes de fármacos tiene (ver gráfica 5). Finalmente, este modelo obtuvo una exactitud del 57.86% superando la obtenida con el modelo sin realizar transferencia de aprendizaje (ver 3.2)
3.6. Resultados con base de datos 2D con aumentación de datos
En este modelo se uso una tasa de aprendizaje de 0.001 Los resultados de la clasificación se muestran en la Fig. 11.
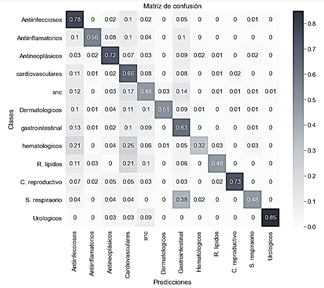
Fuente: Elaborado por los autores.
Figura 11 Matriz de confusión con transferencia de aprendizaje (ResNet50) con imágenes 2D más aumentación digital.
En este modelo la clase de los medicamentos para tratar problemas hematológicos fue la más difícil de predecir. El 25% de los medicamentos usados para tratar problemas hematológicos se predijeron incorrectamente como medicamentos para tratar problemas cardiovasculares. Finalmente, el modelo obtuvo una exactitud del 65.09%, superando a la obtenida con el mismo modelo, pero sin realizar aumentación de datos.
3.7. Resultados con base de datos 3D
En este tercer modelo se uso una tasa de aprendizaje de 0.001 los resultados de la clasificación se muestran en la Fig. 12.

Fuente: Elaborado por los autores.
Figura 12 Matriz de confusión con transferencia de aprendizaje (ResNet50) con imágenes 3D.
Con este modelo se obtuvo una exactitud del 67.47%, superando el obtenido cuando se usó imágenes 2D.
En los resultados obtenidos con la base de datos 2D sin aumentación de datos (grafico 10) los medicamentos que pertenecen a la clase del sistema respiratorio y urológicos no clasificaron ningún medicamento, estas clases son las que menos muestras tienen y esto puede ser un factor que esté causando la baja precisión en las clases con este tipo de muestras. La clase más fácil de predecir fue la de los medicamentos antineoplasticos con un 77% de precisión, por otro, lado, la clase más difícil de predecir (omitiendo al sistema respiratorio y urológicos) fue los medicamentos dermatológicos, el 33% de los medicamentos usados para tratar estos problemas se predijeron incorrectamente como medicamentos para tratar enfermedades del sistema nervioso central y un 22% se predijeron incorrectamente como medicamentos para tratar problemas cardiovasculares.
En los resultados obtenidos con base de datos 2D con aumentación de datos (grafico 11) el modelo obtuvo un aumento en la exactitud con respecto al modelo 3.5. La clase de los medicamentos para tratar problemas hematológicos fue la más difícil de predecir, el 25% de los medicamentos usados para tratar problemas hematológicos se predijeron incorrectamente como medicamentos para tratar problemas cardiovasculares. la clase de los medicamentos urológicos a pesar tener la menor cantidad de fármacos a entrenar dentro de las clases fue la que mejor predijo seguida de la clase de los medicamentos antiinfecciosos.
En los resultados obtenidos con base de datos 3D (grafico 12) la precisión por clase para predecir los medicamentos de uso terapéutico tuvo un aumento en comparación con el modelo con imágenes 2D (grafico 10) y el modelo con imágenes 2D más el aumento de imágenes digitales (grafico 11) en la mayoría de las clases, siendo este uno de los mejores modelos encontrados para predecir la función de los medicamentos cuando tenemos doce clases. Por otro lado, la clase más difícil de predecir fue los medicamentos urológicos, el 38% de los fármacos usados para tratar estos problemas se predijeron incorrectamente como medicamentos para tratar problemas antineoplasticos finalmente la clase que mejor predijo fue los medicamentos antiinfecciosos con un 86% de precisión.
3.8. Validación de los modelos con la desloratadina
Para verificar si efectivamente los modelos entrenados con vistas 2D de la estructura 3D de las moléculas, aportan una mejor información sobre el uso terapéutico de un fármaco; nosotros hemos validado el modelo con la Desloratadina. Este medicamento fue creado inicialmente para uso Antihistaminico (inhibe ampliamente la respuesta alérgica), pero el trabajo de Carretero [31] demostró que también posee un uso Antiinflamatorio.
El uso terapéutico Antihistamínico no se incluyó dentro de las clases escogidas para el entrenamiento. Es así como, los modelos a comparar deberían predecir que la Desloratadina tiene un uso terapéutico principalmente Antiinflamatorio. Se debe aclarar, que la Desloratadina no fue usada en el entrenamiento de ninguno de los modelos, por lo tanto, las redes a validar no han memorizado su estructura molecular.
3.9. Validación del modelo con imágenes 2D
En la Fig. 13 se muestra la imagen 2D usada para predecir el uso terapéutico de la Desloratadina con el modelo entrenado con imágenes 2D más aumentación de datos. El resultado obtenido se muestra en la Fig. 14.

Fuente: National Library of Medicine (NLM).
Figura 13 Imagen 2D de la estructura química de la Desloratadina, fuente Puchem.

Fuente: Elaborado por los autores.
Figura 14 Predicción del uso terapéutico de la Desloratadina usando el modelo entrenado con imágenes 2D (Valores de Precisión).
Los valores de la precisión para el top-3 del uso terapéutico obtenido de la Desloratadina se muestran en la Tabla 2.

3.10. Validación del modelo con imágenes 3D
En la Fig. 15 se muestran las seis vistas 2D usada para predecir el uso terapéutico de la Desloratadina con el modelo entrenado con imágenes 3D. Con cada una de las seis vistas el modelo predijo el uso terapéutico de la Desloratadina como un medicamento que ayuda en los procesos antiinflamatorios. El resultado obtenido con la vista 1, se muestra en la Fig. 16, las gráficas son similares para las demás vistas ya que los valores de la probabilidad para el Antiinflamatorio fueron superiores al 99%. Los valores de la precisión para el top-3 del uso terapéutico obtenido de la Desloratadina con cada una de las 6 vistas, se muestran en la Tabla 3.

Fuente: Elaborado por los autores.
Figura 15 Vistas 2D de la estructura química 3D de la Desloratadina.

Fuente: Elaborado por los autores.
Figura 16 Predicción del uso terapéutico de la Desloratadina usando el modelo entrenado con vistas de la imagen 3D (Valores de Precisión).
Tabla 3 Top-3 de la predicción del uso terapéutico de la Desloratadina usando el modelo entrenado con imágenes 3D.

Fuente: Elaborado por los autores.
Observamos en la Tabla 2 que entrenando el modelo solamente con imágenes 2D de la estructura molecular, el modelo no predice que la Desloratadina tiene un principio activo asociado con un uso Antiinflamatorio. Por el contrario, los resultados mostrados en la Tabla 3, muestran que entrenando el modelo con vistas de la imagen 3D de la estructura molecular, el modelo predice correctamente que la Desloratadina tiene un principio activo asociado con un uso Antiinflamatorio.
4. Discusión
En este trabajo exploramos la posibilidad de utilizar técnicas de aprendizaje profundo para predecir el uso terapéutico de fármacos, basándose únicamente en la información tridimensional de su estructura molecular. Para la predicción se utilizó una red neuronal convolucional, entrenada con seis vistas de la imagen tridimensional de la estructura molecular de cada fármaco. A pesar de que únicamente se usaron 6 vistas, los resultados fueron superiores que con el entrenamiento realizado solamente con la información bidimensional de la estructura molecular del fármaco.
Cuando se usó transferencia de aprendizaje, el modelo entrenado con la información 2D solo mejoró la exactitud un 2.5% respecto al modelo entrenado con la información 2D aumentada digitalmente. Sin embargo, el modelo entrenado con la información 3D de la estructura molecular (6 vistas) mejoró la exactitud en un 10% respecto al modelo entrenado con la información 2D.
Finalmente, mostramos con la predicción del uso farmacéutico de la Desloratadina, que estos modelos pueden ser utilizados para la clasificación de medicamentos con múltiples usos terapéuticos. De esta manera, es posible reinterpretar los resultados de la clasificación. Por ejemplo, los fármacos clasificados de forma incorrecta para una determinada clase podrían ser una indicación de su potencial para un nuevo uso terapéutico. Por lo tanto, una clasificación errónea puede dar lugar a descubrimientos inesperados. Este enfoque abre una gran vía para la aplicación de los modelos de redes neuronales convolucionales en el campo de la reorientación de medicamentos.
Para trabajos futuros se puede tener en cuenta las siguientes recomendaciones:
Aumentar la base de datos con la información 3D de la estructura molecular. La tridimensionalidad de las estructuras moleculares permite que se puedan tomar una mayor cantidad de vistas que las utilizadas en este trabajo por razones de capacidad de computo. De esta manera, se podría balancear la base de datos nivelando las clases que tengan menos fármacos.
Optimizar la estructura molecular de los fármacos de tal forma que su representación 3D sea la más adecuada. Se pueden explorar técnicas de optimización geométrica, cuyo objetivo es obtener las conformaciones estructurales de más baja energía para predecir la disposición tridimensional óptima de los átomos en una molécula.

