











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkEl concepto de biodisponibilidad fue acuñado en 1945 por Oser, et al. 1, para indicar el tiempo y la fracción que un fármaco requiere para alcanzar la circulación general, y la velocidad con que esto ocurre, mediciones que los autores hicieron originalmente con vitaminas en solución y en tabletas 2,3. En Colombia, la definición oficial de ‘biodisponibilidad’ incluida en la Resolución 1124 de 2016 4, alude a la tasa y la extensión con las que un principio activo es absorbido y llega a la circulación general.
Dos medicamentos se consideran equivalentes farmacéuticos si contienen la misma cantidad molar de principio activo con la misma forma farmacéutica y vía de administración y, además, si cumplen con los estándares de comparación. Se denominan alternativas farmacéuticas a aquellos productos que, conteniendo el mismo principio activo, pueden diferir en la forma de dosificación, la concentración o la especie química del ingrediente activo. Se considera que dos productos farmacéuticos son bioequivalentes si, siendo equivalentes o alternativas farmacéuticas, su biodisponibilidad en términos de velocidad y magnitud de la absorción después de la administración de la misma dosis molar en las mismas condiciones, es similar en un grado tal que se puede esperar que sus efectos, en lo esencial, también lo sean 4,5. De esta manera, es posible confirmar la equivalencia terapéutica y establecer la intercambiabilidad entre el producto ‘multifuente’ y el producto de referencia, garantizar la seguridad y eficacia de los productos farmacéuticos, y asegurar el acceso a los medicamentos 6.
La demostración de la bioequivalencia o la equivalencia terapéutica entre productos farmacéuticos puede hacerse empleando métodos in vivo e in vitro. Los primeros son idóneos cuando hay un riesgo de que las posibles diferencias en la bioequivalencia puedan resultar en la falta de equivalencia terapéutica, e incluyen estudios comparativos farmacocinéticos, farmacodinámicos y ensayos clínicos 7. Por otra parte, en los ensayos in vitro se comparan los perfiles de disolución de un producto de referencia, generalmente el innovador, y uno multifuente, lo que puede ser suficiente para establecer la equivalencia en determinados casos.
Cabe resaltar que los ensayos clínicos comparativos son los menos utilizados debido a que, por lo general, requieren un gran número de pacientes, tienden a ser poco sensibles frente a las diferencias en la formulación, son costosos y, eventualmente, poco éticos. No obstante, aunque los estudios de biodisponibilidad in vivo son los métodos de elección para demostrar la bioequivalencia de productos administrados por vía oral 4, los estudios in vitro, ajustados a los lineamientos del Sistema de Clasificación Biofarmacéutica (SCB), son una alternativa práctica y económica para evaluar la intercambiabilidad 8.
Una de las categorías de medicamentos de mayor consumo en Colombia, son los analgésicos no narcóticos y los antipiréticos 9,10, los cuales incluyen los denominados analgésicos antiinflamatorios no esteroides 11. La mayoría es de venta libre, lo que determina el tipo de publicidad y los canales de comercialización, dejando al criterio del paciente la elección del medicamento.
Los principios activos de primera elección de los pacientes son, en su orden, el diclofenaco, el ibuprofeno y el piroxicam. El primero de estos es el que más se consume, por lo cual se eligió para comparar la disolución de las diversas marcas de tabletas disponibles en el mercado colombiano 12. Asimismo, el hecho de que el diclofenaco sódico pertenezca a la clase II del SCB (baja solubilidad, alta permeabilidad) 13, representa un reto para los formuladores, que deben orientar sus esfuerzos a reducir el tiempo de disolución para lograr el mejor resultado terapéutico.
Materiales y métodos
Materiales
Se utilizaron el acetato de sodio trihidrato, el ácido acético, el ácido clorhídrico, el fosfato monobásico de potasio y acetonitrilo de Merck®, el hidróxido de sodio de Panreac®, y el diclofenaco sódico estándar primario de Sigma-Aldrich®, los cuales se mantuvieron almacenados bajo rigurosas condiciones de control de temperatura (20 ºC) y humedad (40 %).
Equipos e instrumentos
Se emplearon los siguientes equipos: equipo de disolución Sotax™ (Allschwil, Suiza); potenciómetro- pH Beckman 50™ (Fullerton, CA, USA); balanza analítica con sensibilidad de 0,1 mg de Mettler Toledo AB204™ (Greifensee, Suiza); calibrador digital Mitutoyo™ (São Paulo, Brasil); espectrofotómetro UV-VIS Spectroquant™ Merck (Darmstadt, Alemania); durómetro Kraemer HC 6.2™ (Darmstadt, Alemania); equipo de desintegración Copley™, serie DTG 2000 (Nottingham, UK); baño ultrasonido Elma E30H™ (Hohentwiel, Alemania), y cromatógrafo HPLC, modelo V2100™, Merck-Hitachi (Tokio, Japón).
Muestras
Se adquirieron tabletas de 50 mg de diclofenaco sódico con revestimiento entérico, distribuidas por ocho marcas en los establecimientos comerciales farmacéuticos de Bogotá. Las tabletas adquiridas se identificaron de manera aleatoria con letras de la A a la H. Entre las marcas, se contaba el producto innovador (F) y otros siete de denominación genérica. De cada marca evaluada, se obtuvieron dos lotes identificados como 1 y 2, con 60 tabletas cada uno.
Validación de la metodología analítica para la cuantificación
La validación de la metodología se hizo en términos de selectividad, linealidad, precisión, exactitud y límite de cuantificación, según las especificaciones de la edición 39 de la United States Pharmacopeia (USP 39) 14 para ensayos de categoría III y de la guía de validación de procedimientos analíticos Q2 (R1) del International Committee for Harmonization (ICH) 15, empleando espectrofotometría UV-VIS.
Para el estudio de la selectividad, se tuvieron en cuenta la variedad de sustancias auxiliares de formulación empleada comúnmente en la elaboración de las formas sólidas del diclofenaco 13 con revestimiento entérico, entre las que se cuentan la croscarmelosa de sodio, la celulosa microcristalina, la povidona K29, la lactosa, el estearato de magnesio, el polisorbato 80, el almidón de maíz y el Eudragit® L-30D y L-100. En el cuadro 1 se presentan los criterios de aceptación para la validación del método analítico.
Cuadro 1 Evaluación de las características de desempeño de la metodología

r: coeficiente de correlación; r2 : coeficiente de determinación; t: prueba tv de Student (α=0,05; gl=n-2); ta: t para intercepto; tb: t para pendiente; tr: t para la regresión; tc: t calculado; F: prueba de Fisher (α=0,05); Fc: F calculado; CV: coeficiente de variación; R: recuperación (%); tR: t para la recuperación
Valoración del principio activo
La valoración del principio activo se hizo en un cromatógrafo HPLC con detector UV, según la monografía oficial de la USP 39 para el diclofenaco sódico en tabletas de liberación retardada.
Dimensiones
Se determinaron las dimensiones de diez tabletas de cada lote con ayuda del calibrador digital de Vernier, y se calcularon el promedio y la desviación estándar.
Desintegración
Esta se desarrolló según el capítulo <701> de la USP: se colocaron seis tabletas de un mismo lote, una por cada celda de la canastilla del equipo de desintegración, y como medio de inmersión se empleó inicialmente el ácido clorhídrico 0,1 N, y en la segunda etapa, una solución amortiguadora de fosfato con pH de 6,8 a una temperatura de 37±2ºC. Se registró el tiempo que tardó cada tableta en desintegrarse en su totalidad, y se determinó el tiempo promedio de desintegración para cada lote y cada producto.
Dureza
La prueba se desarrolló según las indicaciones de la monografía <1217> de la USP. Se tomó una muestra de seis tabletas de cada lote, y se colocaron una por una en un durómetro digital. Se midió la fuerza (kgf) necesaria para provocar la ruptura, y se calcularon el promedio y la desviación estándar para cada lote y cada marca.
Prueba de disolución
Esta se desarrolló según las indicaciones de la monografía de la USP 39 para el diclofenaco en tabletas de liberación retardada: aparato II (paletas), velocidad de rotación de 50 rpm y, como medios de disolución, ácido clorhídrico 0,1 N (900 ml) a 37±0,5 °C durante dos horas en la primera fase, y en la segunda, solución tampón de fosfato de potasio con pH de 6,8 durante 45 minutos a la misma temperatura. Al final de los respectivos tiempos, se tomaron alícuotas y, tras las diluciones necesarias en el mismo medio, se hicieron las lecturas de espectroscopia UV-VIS a 260 nm, comparando los resultados con las curvas de calibración construidas en los respectivos medios de disolución. Las mismas tabletas de la primera fase se emplearon en la segunda.
Perfiles de disolución
Los perfiles de disolución se determinaron en tres niveles de pH, usando como medios de disolución ácido clorhídrico con pH de 1,2, y soluciones tampón de acetato de sodio con pH de 4,5 y de fosfato de potasio con pH de 6,8 a 50 rpm, según las condiciones experimentales establecidas para el ensayo de disolución en el anexo técnico 1 de la Resolución 1124 del Ministerio de Salud 4; se tomaron alícuotas a los 5, 10, 15, 30, 45, 60 y 90 minutos. Se evaluaron 12 unidades de dosificación por cada lote, tomando alícuotas de 6 ml de los vasos de disolución y sin reemplazar el volumen muestreado para evitar cualquier interferencia en el medio de disolución. Al igual que en la prueba de disolución, las curvas de calibración se construyeron con un estándar secundario, disuelto en cada uno de los tres valores de pH, y las lecturas se hicieron en la misma longitud de onda (260 nm).
Análisis estadístico
Los datos se organizaron y analizaron matemáticamente, empleando el programa MS Excel 365. Los análisis estadísticos se hicieron con el programa GraphPad Prism, V. 5.00. Los perfiles de disolución se compararon mediante modelos matemáticos independientes 16 basados en el cálculo de los factores de diferencia f1 y de similitud f2 17. Además, se calculó el área bajo la curva por el método de los trapecios (ABC) 18 y, a partir de dichos valores, se calcularon la eficiencia de la disolución 19-21 y el tiempo medio de disolución 22,23.
Resultados
Validación de la metodología analítica
Los resultados de la validación de la metodología analítica en los diferentes medios de disolución (ácido clorhídrico, pH 1,2; acetato de sodio, pH 4,5; y fosfato de potasio, pH 6,8), se presentan en el cuadro1.
La selectividad se evaluó con respecto a los excipientes y los productos generados por la degradación forzada del activo en los tres valores de pH del estudio (1,2, 4,5 y 6,8). El análisis cromatográfico de las muestras permitió concluir que, ni los excipientes de la matriz ni los productos de degradación, generaron señales que pudieran interferir con las correspondientes al diclofenaco.
Como se muestra en el cuadro 1, la prueba t con un nivel de confianza de 95 % y n-2 grados de libertad indicó que, estadísticamente, había proporcionalidad entre la concentración y la reacción analítica para el rango comprendido entre los 27,5 y 82,5 μg/ml, tanto para el sistema como para el método. Asimismo, se evidenció que la pendiente era estadísticamente diferente a cero y que el intercepto no lo era. Por otro lado, la prueba F corroboró que, para el modelo lineal propuesto, los errores sistemáticos no afectaban la linealidad.
En cuanto a la precisión de la metodología, se observó que los coeficientes de variación eran inferiores al 2 %, es decir, que los resultados eran confiables en cuanto a la posibilidad de replicarlos. El mismo resultado se observó con los datos obtenidos en días diferentes (precisión intermedia).
Los porcentajes de recuperación para los tres niveles de pH evaluados, fueron: de 99,0 a 101,2 % con pH 1,2; de 98,4 a 100,4 % con pH 4,5, y 99,5 a 101,2 % con pH 6,8. Con la prueba t de Student se encontró que con ninguno de estos valores existía una diferencia significativa entre la recuperación promedio y el 100 %. Por lo tanto, el método tenía la exactitud requerida para el estudio.
Pruebas de control de calidad
Los resultados obtenidos en las pruebas de control de calidad para los dos lotes de los ocho productos evaluados, se presentan en el cuadro 2.
Cuadro 2 Resultados de las pruebas de control de calidad de las tabletas de diclofenaco sódico de 50 mg

Q: porcentaje de disolución del fármaco declarado en la etiqueta. Los valores corresponden al promedio ± desviación estándar.
‡Lotes 1 y 2, estadísticamente diferentes (p<0,05; n=10); *Lotes 1 y 2, estadísticamente diferentes (p<0,05; n=6)
¥Lotes 1 y 2,estadísticamente diferentes (p<0,05; n=12)
Los productos evaluados eran tabletas circulares, lisas y uniformes, con diámetro promedio de 6,67 a 8,96 mm y altura promedio de 3,53 a 4,33 mm. El peso promedio osciló entre 139,6 y 248,1 mg, lo cual indicaba que el porcentaje de principio activo en la composición estaba entre 20 y 36 % del peso total de las tabletas. Por otro lado, los valores de dureza oscilaron entre 7,3 y 21,9 kgf. En cuanto a los tiempos de desintegración, ninguno de los lotes de los ocho productos se desintegró en el medio ácido, en tanto que, con pH 6,8, todos los lotes lo hicieron en menos de 30 minutos.
Según la cuantificación del contenido, todos los productos se encontraban dentro del rango de 90 a 110 % establecido en la monografía sobre diclofenaco de la USP 39, con excepción del lote 2 de los productos D y G, los cuales se situaron por encima del límite superior, con valores de 110,97 % y 112,87 %, respectivamente. En cuanto a la prueba de disolución en fosfato, la farmacopea establece que, con pH 1,2, no tendría que disolverse más del 10 % del contenido declarado en 120 minutos y, con pH 6,8, tendría que hacerlo, como mínimo, un 75 % del contenido declarado en 45 minutos. Se encontró que todos los lotes de los ocho productos cumplían con la especificación en medio ácido; sin embargo, con pH 6,8, el lote 2 del producto E, y los lotes 1 y 2 del producto G, presentaron valores inferiores al mínimo exigido a los 45 minutos.
Es importante resaltar que las pruebas de disolución para el control de calidad tienen dos objetivos principales: verificar la reproducibilidad de los lotes según las especificaciones y detectar desviaciones de fabricación 24. Los resultados obtenidos en este ensayo sugieren que los productos en mención pueden presentar diferencias en la formulación o en el proceso de manufactura.
Los perfiles de disolución se desarrollaron según las indicaciones de la Resolución 1124 4 con valores de pH 1,2, 4,5 y 6,8 durante 90 minutos en cada medio, en el aparato de disolución II a 50 rpm, velocidad de agitación con la que se logran mejores resolución y escrutinio 25 (cuadro 3, 4, 5).
Cuadro 3 Porcentaje promedio de disolución de tabletas de diclofenaco sódico de 50 mg con pH 1,2

Q: porcentaje de disolución del fármaco declarado en la etiqueta. Cada valor corresponde al promedio ± desviación estándar (n=12).
Cuadro 4 Porcentaje promedio de disolución de las tabletas de diclofenaco sódico de 50 mg con pH 4,5
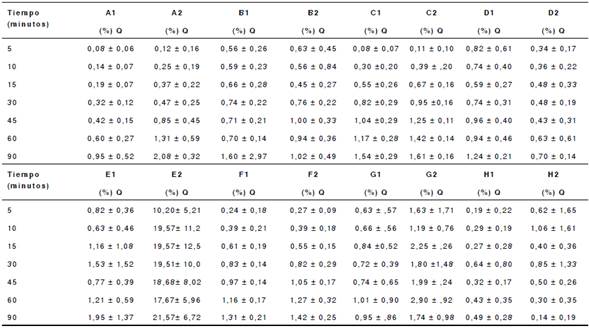
Q: porcentaje de disolución del fármaco declarado en la etiqueta.
Cada valor corresponde al promedio ± desviación estándar (n=12).
Cuadro 5 Porcentaje promedio de disolución de tabletas de diclofenaco sódico de 50 mg con pH de 6,8

Q: porcentaje de disolución del fármaco declarado en la etiqueta.
Cada valor corresponde al promedio ± desviación estándar (n=12).
Las celdas sombreadas indican el momento en que se alcanzó el valor de disolución de Q=85 %.
Con pH 1,2 y 4,5, se evidenciaron valores de disolución inferiores al 10 %, con excepción del lote 2 del producto E, el cual presentó un porcentaje de disolución de 21,57 % con pH 4,5. Con pH 6,8, los productos A, B, C, F, H, D (lote 2) y E (lote 1), lograron el 85 % de la disolución del fármaco a los 30 minutos de iniciado el ensayo. En los productos D (lote 1), E (lote 2) y G, no se alcanzó el 85 % de la disolución a los 30 minutos del ensayo. En el lote 2 de la marca E, es evidente que no se alcanzó el porcentaje de liberación porque, con pH 4,5, más de 27 % ya había sido liberado.
Para comparar los perfiles de disolución, se emplearon modelos matemáticos independientes, entre ellos, los factores de diferencia f1 y de similitud f2 17, el área bajo la curva (ABC), la eficiencia de la disolución y el tiempo medio de disolución (cuadro 6). Los perfiles de disolución de todas las marcas se compararon con el del producto innovador (F) con pH 6,8, valor con el que se libera el fármaco. Para que dos perfiles de disolución se consideren similares, el factor f1 debe estar entre 0 y 15, y el factor f2, entre 50 y 100 (diferencia menor de 10 %) 26,27. De los productos evaluados, solo el B presentó un valor f1 entre 0 y 15, y un f2 superior a 50.
Cuadro 6 Valores de f1, f2, área bajo la curva (ABC), eficiencia de la disolución (ED) y tiempo medio de disolución (TMD) de los productos A a H. Cada valor corresponde al promedio de 24 unidades. F corresponde al producto de referencia (innovador).

Al comparar los valores de la eficiencia de disolución, se evidenció que los productos A, B, C, E y H presentaron valores iguales o superiores al 80 %.
Los valores de tiempo medio de disolución (TMD) oscilaron entre 10,42 y 30,85 minutos, a partir de los cuales se puede determinar el tiempo promedio requerido para la disolución del fármaco y el efecto de retardo en las presentaciones farmacéuticas con revestimiento entérico 28. Ello permite inferir que algunas marcas pueden emplear uno o varios excipientes que retardan la liberación del fármaco. Este indicador fue muy similar en las marcas F y G.
Discusión
El diclofenaco pertenece a la clase II del SCB 13 (baja solubilidad, alta permeabilidad), es decir que si la disolución es eficiente, la absorción no limita su biodisponibilidad. La normatividad establece que, si el fármaco libera el 85 % o más en 15 minutos (productos de liberación muy rápida), la comparación f2 es innecesaria; sin embargo, se debe contemplar el riesgo de una decisión de intercambiabilidad incorrecta cuando el producto multifuente evaluado es suprabiodisponible. En el cuadro 5 se observa que, con pH 6,8, los productos A, C y H presentaron porcentajes de disolución superiores al 85 % a los 15 minutos 25,29, lo cual sería un criterio para considerar la intercambiabilidad. No obstante, en este caso, dichos productos no deben considerarse equivalentes biofarmacéuticos del producto de referencia, dado que este alcanzó el 85 % a los 30 minutos. Al disolverse de esta forma, las marcas A, C y H tienen una liberación rápida, y son equivalentes e intercambiables entre sí, pero no con el producto F. La conclusión, entonces, es que los productos mencionados son supradisponibles y que la única marca intercambiable con la de referencia sería la B.
En ciertos casos, la condición de supradisponibilidad puede asociarse con la toxicidad dada la llegada masiva del fármaco a la biofase. Esto es poco probable en el caso del diclofenaco: primero, porque su biodisponibilidad es de alrededor del 60 % por la extensión del primer paso; segundo, porque tiene un tiempo de vida media corto, de cerca de una hora 29, y tercero, porque se administra cada 12 horas. Además, su farmacocinética es lineal 13, y en el mercado se ofrecen tabletas de 75 mg. Los resultados sugieren que, si los ensayos de los perfiles de disolución se hubiesen desarrollado a 75 rpm, todas las marcas, con la posible excepción de la G, serían equivalentes biofarmacéuticos. Debe hacerse énfasis en que se trata de equivalencia biofarmacéutica (in vitro) y no de bioequivalencia, pues siendo el diclofenaco de clase II según el SCB, se requieren estudios in vivo para demostrar esta última 30.
Por otra parte, se quiso establecer si había alguna correlación entre dureza, desintegración y eficiencia de la disolución en este fármaco. La teoría básica enseña que, cuanto mayores sean los valores de dureza, mayor es el tiempo de desintegración y menores los porcentajes de disolución 24. La figura 1 muestra esquemáticamente la relación entre estas variables. Si se cumple lo anterior, se esperaría un incremento en los tiempos de desintegración a medida que aumenta la dureza de las tabletas, y una disminución de la eficiencia de disolución, cosa que no ocurrió.
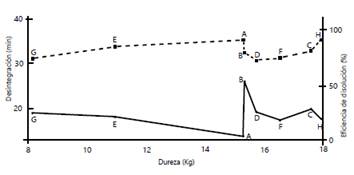
Figura 1 Descripción esquemática de la dureza (eje X), la desintegración (línea sólida, eje Y izquierda) y la eficiencia de la disolución (línea punteada, eje Y derecha) de las tabletas de diclofenaco de 50 mg
Por ejemplo, la marca A tuvo una de las durezas más altas, pero el más bajo tiempo de desintegración, y la mayor eficiencia de disolución, en tanto que la marca B, cuya dureza es igual a la de la marca A, registró el mayor tiempo de desintegración, pero una eficacia de disolución comparable con la del producto de referencia.
En investigaciones previas se había llegado a la misma conclusión 18,19.
El presente estudio permitió validar la metodología analítica para la cuantificación del principio activo del diclofenaco sódico, empleando espectrofotometría UV en pruebas de disolución in vitro, y comprobar el cumplimiento de los parámetros de especificidad, linealidad, precisión, exactitud y límite de cuantificación en tres medios de disolución con pH 1,2, 4,5 y 6,8. Las pruebas de control de calidad del producto de referencia, así como de los productos multifuente, permitieron establecer que los productos D y E no cumplieron cabalmente con las pruebas de valoración del principio activo y del porcentaje de disolución especificadas en la farmacopea vigente, y que el producto G fue el único que no cumplió con ninguna de las pruebas mencionadas.
La comparación de los perfiles de disolución empleando el modelo independiente, permitió demostrar la intercambiabilidad in vitro del producto B y del producto de referencia F. Las marcas A, C y H fueron supradisponibles comparadas con la F, pero esta condición no supone que el fármaco represente un peligro. Además, dadas las propiedades farmacocinéticas del diclofenaco, estas marcas también podrían ser intercambiables con la F.

