











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink1. Introduction
Alzheimer’s is a progressive fatal brain disease that leads to a mental disrepair translated into Dementia. Nowadays, Alzheimer’s is responsible for most of the reported dementia cases, with an incidence of 60 to 80%. Alzheimer’s leads to an increasing loss of memory, cognition and daily living normal function [1]. According to the founds of World Alzheimer Report 2015 [2], 46.8 million people lives with dementia: 9.4 in América, 4.0 in Africa, 10.5 in Europe and 22.9 in Asia; and this value tends to duplicate every 20 years. The 58% of Alzheimer’s cases show up in middle and low-income countries and this percentage is expected to increase to 68% in 2050. The Alzheimer’s world cost is around 818 billion dollars and is thought to be 2 trillion in 2030. If dementia were a country, it would rank 18th in the list of the world’s biggest economies.
There are many manifestations of Alzheimer’s, but probably the most notorious in the progression of the disease are the changes in the metabolism of amyloid β peptide (Aβ), preceding clinical symptoms [3], which basically consist in the accumulation of extracellular deposits called amyloid plaques, composed of Aβ protein aggregates of 38 to 43 amino-acids. The amyloid cascade hypothesis marks that cerebral accumulation of Aβ is a crucial player in the pathogenesis of the disease, due to the initiation of a neurotoxic cascade that leads to further neuronal death. The pharmacological treatments for AD can be symptomatic or etiology based. Symptomatic treatments include acetylcholinesterase inhibitors and N- methyl-D-aspartate (NMDA) receptor antagonists. In contrast, etiology-based treatments include secretase inhibitors, amyloid binders, and tau therapies. Alzheimer´s disease prevention is based on maintaining good daily habits and lifestyle interventions such as exercise, mental challenges, socialization, caloric restriction and a healthy diet [4].
The enzyme responsible for catalyzing the initial cleavage of the amyloid precursor protein (APP) to generate Aβ is β-secretase 1, widely known as β-site amyloid precursor protein cleaving enzyme 1 (BACE1). Genetic alterations in APP are related to early onset of AD, implicating that the amyloid pathway and non-regularities present in Aβ production are critical in the disease analysis [5, 6]. Given the role of BACE1 in Alzheimer’s, its inhibition is thought as a possible blocker of one of the earliest pathological events in the disease, expecting benefits to the patients [3, 5, 6]. Therefore, understanding the dynamics of BACE1 and its homologue BACE2 [7] molecules helps the structural analysis and the study of molecular interaction, opening the way to inhibition or production regulation of the main precursor in Alzheimer’s disease.
The role of cholesterol as a susceptibility factor or a protective agent in neurodegeneration and the link to amyloid-induced cytotoxicity is still controversial. The presence of cholesterol in the cell membrane can modulate conformational changes of specific protein/peptides and a higher membrane rigidity following increased cholesterol content can be protective against aggregate interaction with cell membranes, and aggregate cytotoxicity following perturbation of membrane integrity. Epidemiological studies seem to prove that hypercholesterolemia is a risk factor for AD, but the relation between hypercholesterolemia and brain cholesterol levels is not yet ascertained. The ApoE4 genotype is a prevalent genetic risk factor for AD and the function of ApoE as main cholesterol carrier in the brain also underlies a close cholesterol load-AD risk relation. In addition, cell biology evidences support a critical involvement of lipid raft cholesterol in the modulation of β and γ-secretase cleavage of APP with altered Aβ production [8].
The proper development of efficient treatments is often limited by the lack of knowledge about the mechanism of fibril formation and protein aggregation in AD. One important reason to this lack is the difficulty to observe atomic details and aggregation process in proteins by using traditional molecular biology and crystallography. Nevertheless, molecular dynamics (MD) simulations allows the study of detailed pathway of protein aggregation and provide explanations for experimental results [9], employing either the Newton’s law or more comprehensive approaches such as the Langevin equations to analyze the motion of simple and complex molecules to properly simulate biological environments [10], for example. Structural motions depending on solute or temperature are important to identify the pattern of ligand-protein or protein-protein complexes. The use of MD simulations in the discovery of new drugs is useful to advise structural cavities required to design novel structures with higher affinity to the target, also to refine the 3D structure of targets. On the other hand, MD simulations improve free energy values for protein ligand recognition that can be validated by experimental procedures [11].
The aim of this work is to perform a MD simulation protocol to understand the dynamics of single and double β-secretase 1 (BACE1) and 2 (BACE2), by a structure evaluation of their transmembrane domains while inserted in simulated neural membrane systems considering different levels in membrane cholesterol content, in order to simulated healthy and Alzheimer disease neural membranes, as there is evidence of the cholesterol levels increased in AD [12]. The ultimate goal is to contribute to the development of etiology-based treatments against AD.
2. Methodology
Structure prediction for BACE1 and BACE2 transmembrane domains was accomplished via the web server PEP-FOLD v3.1 from the corresponding aminoacidic sequence. Energy of resulting models was evaluated with the aid of coarse-grained optimized potential for efficient structure prediction (sOPEP) [10]. This potential is based on a six-bead model per amino acid, and yields very high structural precision of the backbone while discriminating native from ab initio-generated structures of peptides [13]. Model lipid bilayers were built on the CHARMM-GUI web server [14] where a heterogeneous protein/membrane system was implemented. This server is a web‐based graphical user interface of CHARMM (Chemistry at HARvard Macromolecular Mechanics), which is a highly versatile and widely used molecular simulation program developed at Harvard University. The bilayer composition was 92 mol% 1,2-dimyristoyl-sn-glycero-3-phosphocoline (DMPC) and 8 mol% 1,2-dimyristoyl-sn-glycero-3-phospho-L-serine (DMPS) [Figure 1]. This was maintained for both the upper and the lower leaflet [15]. From this basis membrane composition, two types of membranes were generated that contained 10 mol% (CHL10) and 50 mol% (CHL50) cholesterol. This was intended to simulate a normal neural membrane and one under the Alzheimer´s disease, respectively. Simulation domain was a rectangular box with dimensions of 58.15 Å in X and Y directions and 79 Å in the Z direction for a total area along the X and Z direction of 3381 Å2. Transmembrane domains were located along the Z axis. Net charged was achieved by locating 8 cations in the bilayer with the aid of a Monte Carlo approximation. Lipid penetration was checked and the employed force field was CHARMM36. For the double systems, once the first domain was located in the bilayer, the second domain was inserted along the Z axis at a distance of 20 Å with the aid of the software VMD v1.9.3. Minimization and equilibration of MD simulations were conducted in GROningen MAchine for Chemical Simulations (GROMACS) v4.6.7 [16, 17].

3. Results
3.1 TM domains
Peptides of TM domains for both BACE1 and BACE2 were optimized and exhibited alpha helical structures with approximately 5 Å in diameter and 30 Å in length [Figure 1C). The protein data bank (PDB) file of both peptides was recovered for further processing in GROMACS. This file format provides a standard representation for macromolecular structure data derived from X-ray diffraction and NMR studies. Hydro-phobic in the Kyte-Doolittle´s scale is very pronounced as expected for TM domains that are normally located in the hydrophobic core of the lipid bilayer.
3.2 Peptide-Membrane systems
The heterogeneous systems were successfully built [Figure 1A). BACE1 insertion in the lipid bilayer is shown in Figure 1A (white domain in the core) while therein cholesterol is shown in yellow flanking the bilayer.
3.3 MD simulations of single TM domains
Figure 2A shows the Root-mean-square deviations (RMSD) for the single TM domains in the normal and Alzheimer´s lipid bilayers. The recovered traces suggest that allotted time was sufficient for equilibration. Fluctuations in the peptide structure along the MD simulations are shown in Figure 2B where BACE1 appears to oscillate with higher amplitudes around the mean positions compared with BACE2.

3.4 Hydrogen bonding of single TM domains
Figure 3A compares the number of hydrogen bonds along the simulation for BACE1 and BACE2 in the normal and Alzheimer´s lipid bilayers. Figure 3B shows the time progression of hydrogen bonding as a function of residue position in the TM domain.
3.5 Changes in the secondary structure of single TM domains
Secondary structural changes are plotted along the MD simulation for BACE1 and BACE2 in normal and Alzheimer´s lipid bilayers [Figure 4]. For both TM domains substantial changes in the secondary structure are observed as cholesterol was increased from 10 mol% to 50 mol%.


3.6 MD simulations of double TM domains
Figures 5A, 6A, and 7A show the Root-mean-square deviations (RMSD) for the double BACE1/BACE1, BACE1/BACE2 and BACE2/BACE2 TM interacting domains in the normal and Alzheimer´s lipid bilayers, respectively. Fluctuations (RMSF) in the peptide structures along the MD simulations are shown in Figure 5B, 6B and 7B. RMSD and RSMF data were collected for both parallel and antiparallel domain disposition.



3.7 Hydrogen bonding of double TM domains
Figures 8A, 9A and 10A compare the number of hydrogen bonds along the simulation for the double BACE1/BACE1, BACE1/BACE2 and BACE2/BACE2 interacting TM domains in the normal and Alzheimer´s lipid bilayers, respectively. Figures 8B, 9B and 10B show the time progression of hydrogen bonding as a function of residue position in the interacting TM domains.
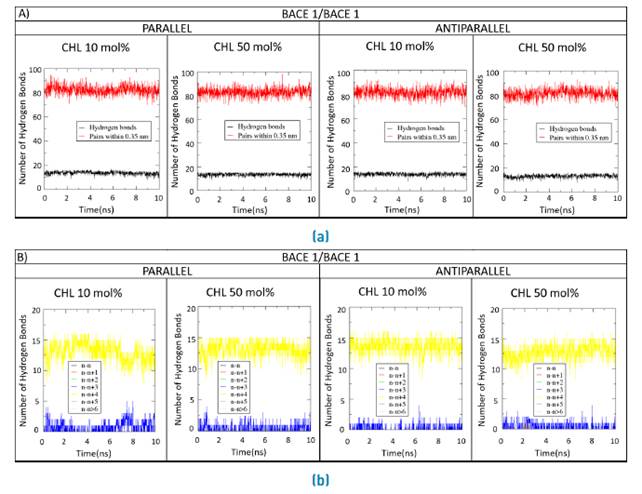
Figure 8 Forming and breaking of H-bonds for the double BACE1/BACE1 systems. a) Parallel and b) Antiparallel number of H-bonds and their constituent amino acid pairs within 0.35 nm of BACE1/BACE1 transmembrane domains during 10ns MD simulations with a molar concentration of membrane cholesterol of either 10%mol or 50%mol
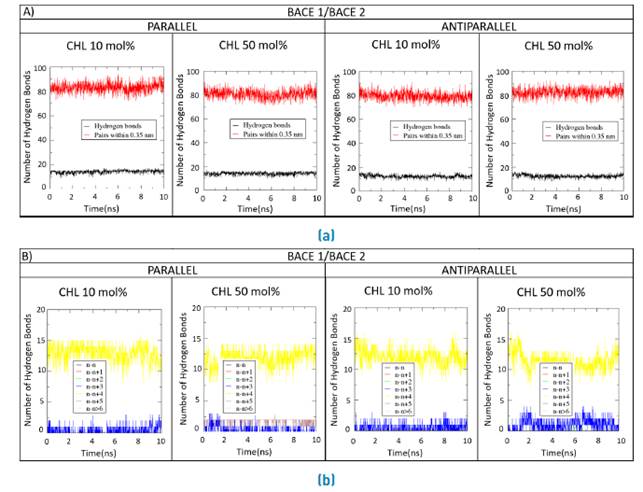
Figure 9 Forming and breaking of H-bonds for the double BACE1/BACE2 systems. a) Parallel and b) Antiparallel number of H-bonds and their constituent amino acid pairs within 0.35 nm of BACE1/BACE2 transmembrane domains during 10ns MD simulations with a molar concentration of membrane cholesterol of either 10%mol or 50%mol
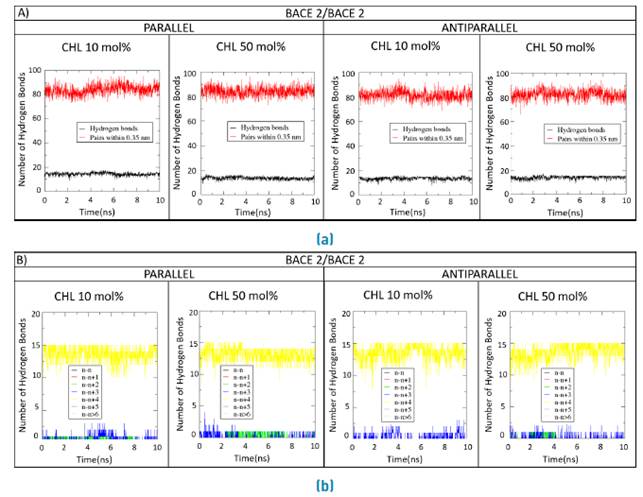
Figure 10 Forming and breaking of H-bonds for the double BACE2/BACE2 systems. a) Parallel and b) Antiparallel number of H-bonds and their constituent amino acid pairs within 0.35 nm of BACE2/BACE2 transmembrane domains during 10ns MD simulations with a molar concentration of membrane cholesterol of either 10%mol or 50%mol
3.8 Changes in the secondary structure of double TM domains
Secondary structural changes are plotted along the MD simulation for the double BACE1/BACE1, BACE1/BACE2 and BACE2/BACE2 interacting TM domains [Figure 11, 12, 13]. Changes in the secondary structure are evaluated for an increase in the cholesterol contents from 10 mol% to 50 mol%.
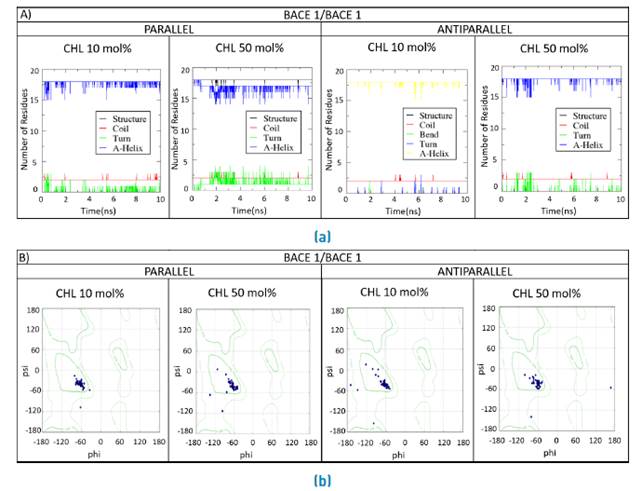
Figure 11 Secondary structure for the double BACE1/BACE1 systems. a) Parallel and b) Antiparallel coils, bends, turns and helices were monitored as time progressed during the MD simulation for the double TM domains in lipid bilayers containing 10 mol% and 50 mol%
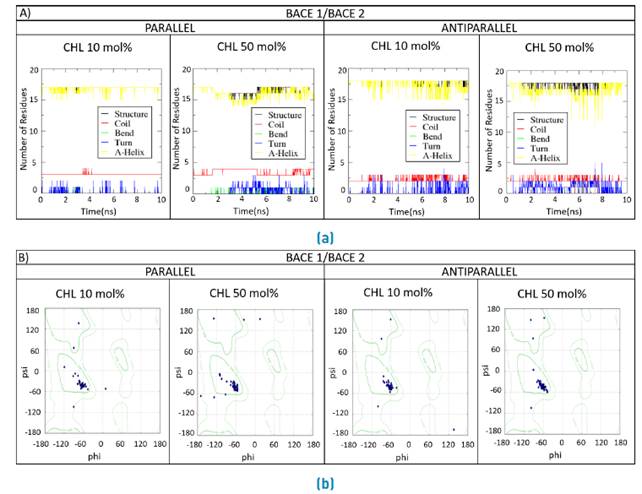
Figure 12 Secondary structure for the double BACE1/BACE2 systems. a) Parallel and b) Antiparallel coils, bends, turns and helices were monitored as time progressed during the MD simulation for the double TM domains in lipid bilayers containing 10 mol% and 50 mol%.

4. Discussion
As evidenced in Figure 2B, fluctuations around the mean value for BACE1 appear considerably higher compared with BACE2. This can be attributed to the presence of methionine and leucine residues in the structure, which were more frequently associated with locations of higher variations in position [18]. The high hydrophobicity of these type of residues (according to the Kyte-Doolittle scale) suggest a much more dynamic interaction along the simulation trace. Nevertheless, this notion should be considered with care considering that leucine and methionine are more abundant in the structure than other residues. Additionally, leucine exhibits a lower molecular size, which confers it the possibly to take upon several conformations along the MD trajectory [6].
Deviations with respect to the standard alpha helical structure are considerably higher for both TM domains as the contents of cholesterol is increased from 10 %mol to 50 %mol. This is supported by the reduction in the number of n-n+4 hydrogen bonds and the concomitant increase of n-n+3 or n-n+5 [Figure 3B). Moreover, more disordered secondary structural features such as 3-10 helices and bends or turns start to dominate the conformational landscape at 50 %mol cholesterol [Figure 4]. This was confirmed by the inversely proportional relationship between the helices radii and lengths as well as a relatively higher RMS deviation with respect to the ideal alpha helix. Even though hydrogen bonding change along the MD trace, the number of bonds remains approximately constant and within a 3.5 Angstrom limit. This strongly suggests that the involved pairs are temporarily replaced by positions nearby along the trajectory. Despite this dynamic evolution of the hydrogen bonding, donor-acceptor distribution seems to approach to an average value of about 3 Angstrom. This is also the case for the angle distribution that remains approximately constant in about 12-13 degrees.
The relatively small fluctuations in secondary structure were confirmed by the Ramachandran plots where final conformations were located within probabilistically allowed phi and psi configurations.
Traces for RMSD for the for the double BACE1/BACE1, BACE1/BACE2 and BACE2/BACE2 TM interacting domains equilibrate within the allotted time frame [Figures 5A, 6A and 7A). As opposed to the single systems, the marked tendency for oscillations at 50 %mol cholesterol was not observed for the double systems [Figures 5B, 6B and 7B).
Some deviations with respect to the standard alpha helix were observed for the double systems in the presence of high cholesterol levels as evidenced by the subtle changes in the donor-acceptor dynamics of hydrogen bonds at 50 %mol cholesterol [Figures 8B, 9B and 10B). Also, the number of hydrogen bonds remain approximately constant along the MD traces, which suggests that bonding occurs between nearest neighbors [Figures 8A, 9A and 10A).
Secondary structural changes were observed for high cholesterol levels as evidenced by the presence of alternative conformations such as pi and 3-10 helices, bends and turns [Figures 11A, 12A y 13A). This was further confirmed by the location of final conformations in the Ramachandran plots [Figures 11B, 12B y 13B). Changes were particularly exacerbated for residues located in close proximity to the termini of sequences. Additionally, as for the single systems, hydrophobic residues such as leucine and phenylalanine appear to fluctuate along the traces thereby suggesting an important role in the interactions with phospholipids.
Finally, the interaction potential between the TM domains appears higher for the BACE2/BACE2 system in the presence of high cholesterol levels and particularly in the antiparallel disposition.
5. Conclusion
The MD simulation protocol implemented here proved useful to explore the interaction potential and secondary conformational changes of the TM domains of BACE1 and BACE2, two important (-secretases thought to be involved in the deregulated signaling cascade leading to Alzheimer´s. According to our findings, high cholesterol levels appear to induce subtle secondary structural changes to single BACE2 TM domains as evidenced by the presence of alternative helical motifs such as 3-10 and pi helices.
Our results strongly suggest that an increase in the cholesterol levels promotes exacerbated lateral interactions for BACE2-BACE2 interacting pairs, which were considerably stronger when TM domains were inserted in an antiparallel manner in the lipid bilayer. Additionally, a subtle disruption in the ideal helical conformation was identified for the interacting pairs with changes in the hydrogen bonding and fluctuations in the more hydrophobic residues along the MD traces.
Taken together our findings provide robust evidence to support the notion that altered bilayer compositions are responsible for deregulation of membrane proteins and receptors via exacerbated lateral interactions that ultimately lead to neurodegenerative conditions and cancer.

