











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCCIÓN
La adrenoleucodistrofia es considerada la enfermedad peroxisomal más frecuente. Esta entidad lleva a una acumulación anormal de ácidos grasos de cadena muy larga (AGCML) en diferentes tejidos, principalmente en las células nerviosas, de la corteza adrenal y de Leydig en los testículos 1. Abarca un determinado grupo de enfermedades con un origen genético definido, ligado al cromosoma X, y una variabilidad fenotípica conocida como el complejo adrenoleucodistrofia/adrenomieloneuropatía (complejo ALD/ AMN), que depende principalmente de la edad de presentación 2. Existen tres presentaciones clínicas principales, la forma cerebral de la infancia, la adrenomieloneuropatía y la insuficiencia adrenal aislada 3. Se presenta un caso de un hombre adulto con múltiples consultas a los servicios de urgencias en relación con síntomas medulares dorsales, con evidencia de insuficiencia suprarrenal y paraclínicos que soportan el diagnóstico de adrenomieloneuropatía.
Presentación de caso
Se trata de un hombre de 32 años, trabajador en una planta de sacrificio de aves. Presenta desde los últimos diez meses debilidad intermitente de las extremidades inferiores, por lo que había requerido hospitalización en algunas ocasiones. Los episodios habían sido manejados con corticoides endovenosos con mejoría clínica significativa.
Nueve días antes de su último ingreso, presenta exacerbación de la debilidad y dolor de características mal definidas e intensidad variable en miembros inferiores, en ausencia de falla del control de esfínteres u otros síntomas neurológicos. Como antecedentes refiere insuficiencia suprarrenal diagnosticada 18 meses atrás, en manejo con prednisona, sin otros antecedentes personales y familiares relevantes. A la revisión por sistemas refiere disfunción eréctil durante los últimos tres meses. En los últimos dos años el paciente ha experimentado pérdida de peso, oscurecimiento de piel, pérdida de cabello y pestañas, astenia, polidipsia, poliuria y nausea intermitente.
Al examen físico se aprecia hiperpigmentación cutánea y mucosa, principalmente en pliegues palmares y plantares, también en encías y labios. Los signos vitales se encuentran en rango de normalidad, con un peso de 74 kg y una talla de 1,72 m. El examen mental y de pares craneales es normal, el examen motor evidencia paresia simétrica 4/5 en extremidades inferiores, hiperreflexia simétrica en extremidades inferiores y Babinski bilateral. Los exámenes sensitivo y cerebeloso son normales, requiere apoyo para la marcha y no presenta signos meníngeos ni movimientos anormales.
Desde el punto de vista clínico, se plantea que el paciente cursa con una mielopatía dorsal recurrente, con un aparente componente inflamatorio, que se maneja con metilprednisolona endovenosa, con mejoría parcial de los sintomas. Los paraclínicos del paciente mostraron hemograma, electrolitos sericos, CK total, niveles de vitamina Β 12, ácido fólico, pruebas de función renal y tiroidea normales, glicemia en ayunas de 118 mg/dl, triglicéridos de 319 mg/dl, colesterol total de 341 mg/dl, HDL de 41 mg/ dl, LDL de 234 mg/dl, VDRL sérico no reactivo, ELISA VIH negativo, anticuerpos contra HTLV 1 y 2 negativos y anticuerpos antinucleares positivos 1:160 diluciones con un control posterior negativo.
Antes del inicio de prednisona se evidenció un cortisol sérico (8:00 a. m.) disminuido en 5,02 mcg/dl (6,2-19.4). La TAC de abdomen y pelvis no mostró lesiones suprarrenales. El análisis de LCR no mostró anormalidades, incluido bandas oligoclonales negativas, neuroconducciones y electromiografía de las cuatro extremidades sin evidencia de anormalidades. La resonancia magnética (RM) de columna dorsal simple y con contraste evidenció hidromielia T6-T8 y la RM cerebral simple y contrastada hiperintensidad en secuencias T2 y FLAIR de los tractos corticoespinales bilateralmente, a nivel de cápsulas internas y en todo su recorrido en el tallo cerebral en relación con cambios por degeneración Walleriana (figuras 1, 2 y 3).
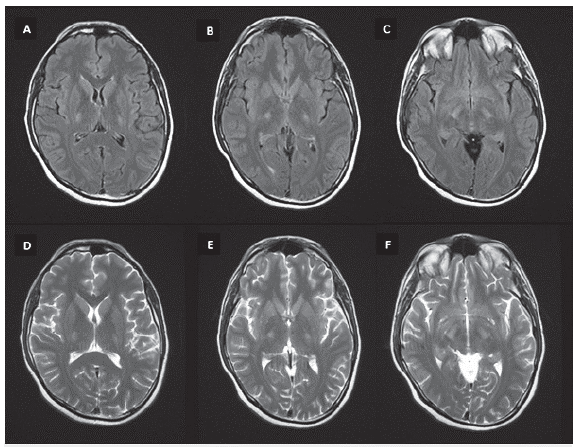
Figura 1 RM cerebral simple, cortes axiales secuencias FLAIR (A, B, C) y T2 (D, E, F) con hiperintensidad bilateral de la vía piramidal en su trascurso por la cápsula interna
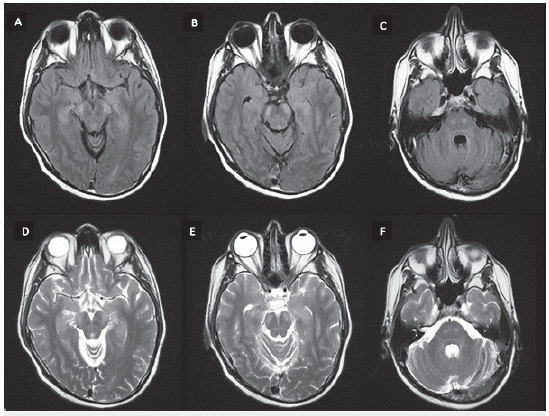
Figura 2 RM cerebral simple, cortes axiales secuencias FLAIR (A, B, C) y T2 (D, E, F) con hiperintensidad bilateral de la vía piramidal a nivel de mesencéfalo y protuberancia
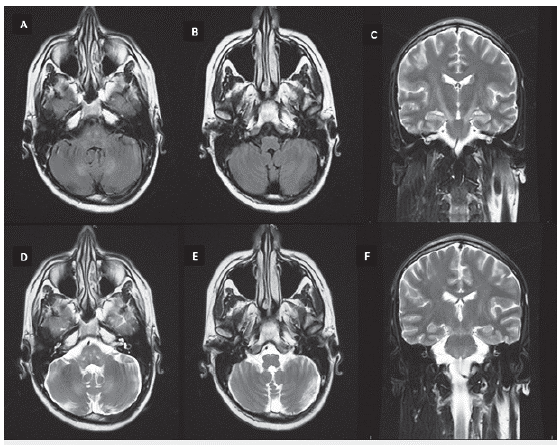
Figura 3 RM cerebral simple, cortes axiales secuencias FLAIR (A, B) y T2 (D, E) con hiperintensidad bilateral de la vía piramidal a nivel de protuberancia y bulbo raquídeo. Cortes coronales secuencia T2 con hiperintensidad de la vía piramidal a nivel de capsulas internas y tallo cerebral (C y F).
El paciente presentó mejoría parcial del déficit neurológico luego del uso de corticoides endovenosos y terapia física, y logró caminar sin apoyo, sin embargo, dos meses después vuelve a consultar por exacerbación de debilidad y dolor neuropático severo en extremidades inferiores que requiere de nuevo uso de corticoides endovenosos y manejo analgésico agresivo, en tanto se continúa el plan de rehabilitación física establecido.
Se obtuvieron resultados de medición de ácidos grasos de cadena muy larga en suero (cromatografía de gases), los cuales son reportados como alterados, con niveles de ácido hexacosanóico 26:0 en 5,89 mcgmol/L (0,22-0,88), ácido docosanóico 22:0 en 124,3 mcgmol/L (51,1-113,4) y ácido tetracosanóico 24:0 en 202,4 mcgmol/L (44,3-92,4), relación 24:00/22:00 1,63 (0,55-0,89) y relación 26:00/22:00 0,05 (0,004-0,021) (tabla 1).
Tabla 1 Resultados de medición de ácidos grasos de cadena muy larga en suero (cromatografía de gases).

Con base en la clínica, hallazgos imagenológicos y de laboratorio se considera el diagnóstico de adrenoleucodis-trofia variedad adrenomieloneuropatía.
DISCUSIÓN
La adrenoleucodistrofia es una enfermedad peroxisomal, caracterizada por un defecto genético ligado a X que impide la betaoxidación de los ácidos grasos de cadena muy larga y que produce su consecuente acumulación anormal en múltiples tejidos, lo que lleva a diferentes manifestaciones clínicas de variable severidad. El defecto genético consiste en mutaciones en el gen (ABCD1) que codifica para la el transportador de ABC (ATP-binding cassette) en el cromosoma Xq28, este transportador ayuda a formar el canal por el cual los ácidos grados de cadena muy larga entran al peroxisoma para su posterior betaoxidación. Debido a lo anterior, estos AGCML no se betaoxidan, se acumulan en los tejidos y generan disfunción celular de manera directa o a través de unas respuestas inmunes (humoral y celular), principalmente en células del sistema nervioso central, de la corteza suprarrenal y de la células de Leydig del testículo 1,3-5.
La neuropatología de las lesiones en el sistema nervioso central en la forma cerebral evidencia cambios de desmielinización inflamatoria que afectan inicialmente los lóbulos occipitales y parietales y luego se extienden hacia lóbulos temporales y frontales 6. La pérdida axonal puede ser considerable, pero el daño mielínico será mayor. Algunas veces hay compromiso del tallo cerebral, en particular el puente, y la médula espinal es usualmente respetada (a excepción de degeneración bilateral de tractos corticoespinales con evidencia de cambios por atrofia espinal) 7.
La adrenomieloneuropatía (forma adulta) se caracteriza por desmielinización inflamatoria o no inflamatoria y axonopatía degenerativa con compromiso de tractos medulares ascendentes y descendentes, especialmente el fascículo de gracilis y corticoespinal 2.
El espectro clínico en el hombre es diverso, con tres formas principales que son la forma cerebral de la infancia, la adremomieloneuropatía y la insuficiencia suprarrenal aislada. En las mujeres cuando hay manifestaciones su curso clínico es leve y de presentación tardía en relación con los hombres 5.
La forma cerebral suele presentarse en niños varones de entre cuatro y ocho años y corresponde a aproximadamente el 35 % de los casos dentro del complejo ALD/AMN. Se caracteriza, en un comienzo, por dificultades en el aprendizaje, alteraciones comportamentales, con posterior deterioro cognitivo, déficit visual, cuadriparesia y crisis epilépticas (en el 20 %%, aproximadamente) 8. La mayoría tiene insuficiencia suprarrenal y algunos tienen la característica piel hiperpigmentada por exceso de hormona adrenocorticotrófica (ACTH). Es frecuente la rápida progresión con discapacidad severa a los 6-24 meses y muerte entre 5 y 10 años del diagnóstico 4,8.
La adrenomieloneuropatía corresponde, aproximadamente, al 40-45 % de los casos del complejo ALD/AMN, y su manifestación principal es la mielopatía, con debilidad piramidal progresiva (paraparesia espástica), alteración de la marcha, alteración en el control de esfínteres y disfunción eréctil. La disfunción gonadal puede preceder las manifestaciones neurológicas y la mayoría cursan con insuficiencia suprarrenal 8. El compromiso cerebral es raro en el momento del diagnóstico (6 °%); sin embargo, un número considerable desarrolla síntomas cerebrales y lesiones desmielinizantes en resonancia magnética cerebral con el transcurrir de los años, empeorando el pronóstico 9.
La insuficiencia suprarrenal aislada puede corresponder al 8 °% de los casos del complejo ALD/AMN, suele presentarse en niños y adultos jóvenes 10, sus síntomas usuales son vómito inexplicado, debilidad, hiperpigmentación de la piel y, luego, con el trascurso del tiempo, la mayoría desarrolla síntomas de adrenomieloneuropatía 11.
Hay presentaciones clínicas menos frecuentes, como lo son la disfunción cerebelosa progresiva en niños y adolescentes, la cefalea con o sin focalización cerebral, el trastorno neurocognitivo mayor en adultos y la disfunción eréctil aislada en hombres con antecededentes familiares de ALD 8.
Las mujeres pueden ser asintomáticas, pero se ha descrito la aparición de mielopatía, polineuropatía e incontinencia fecal con el pasar de los años. El 88 °% de las mujeres mayores de 60 años portadoras de la adrenoleucodistrofia ligada a X puede desarrollar síntomas de la enfermedad, sin embargo, la intensidad de los síntomas suele ser leve y por lo general no desarrolla compromiso cerebral ni suprarrenal 12.
Si se sospecha del diagnóstico de adrenoleucodistrofia ligada a X en un paciente hombre con síntomas neurológicos, con o sin anormalidades típicas en neuroimágenes, o enfermedad de Addison, la cuantificación de ácidos grasos de cadena muy larga en suero es diagnóstica; sin embago, vale la pena resaltar que pueden ocurrir falsos positivos por hemólisis o causas dietarias. En mujeres con la enfermedad, hasta el 15 °% de la cuantificación es normal, por lo que se recomienda complementar el proceso diagnóstico con el análisis de la mutación genética (ABCD1) 2. El análisis de la mutación genética mencionado en hombres cobra importancia cuando los hallazgos clínicos son dudosos o los valores de AGCML son limítrofes. Se debe evaluar la función adrenal, la cual debe debe incluir la medición de cortisol sérico, ACTH y la prueba de estimulación con ACTH 8. Hoy se discute la utilidad y el beneficio futuro de la medición de C26:0 lisofosfatidilcolina en sangre de recién nacidos como método de tamizaje de la enfermedad 13.
La resonancia magnética cerebral en pacientes con presentaciones cerebrales, usualmente niños, siempre es anormal, con desmielinización que predomina en la región parietooccipital bilateral y esplenio del cuerpo calloso, visible como aumento de la señal en secuencias T2 y FLAIR 2. El realce con el medio de contraste de estas lesiones sugiere progresión clínica 14.
En la forma más frecuente de presentación adulta, la adrenomieloneuropatía, la resonancia magnética cerebral generalmente es normal, pero se pueden evidenciar cambios de axonopatía de la vía corticoespinal en secuencias T2 y Flair, o con imágenes avanzadas como tractografía e imágenes con tensor de difusión.
El diagnóstico diferencial imagenológico de estas hiperintensidades de los tractos corticoespinales más común es la esclerosis lateral amiotrófica y la esclerosis lateral primaria, con casos también descritos en cirrosis, encefalopatía hepática y enfermedad de Charcot-Marie-Tooth ligada a X 15,16.
La resonancia magnética de la medula espinal puede llegar a mostrar atrofia inespecífica del cordón medular, pero sin lesiones desmielinizantes o realce con el medio de contraste como en esclerosis múltiple; las imágenes con transferencia de magnetización y tensor de difusión con resonancia pueden detectar anormalidades de los cordones laterales y posteriores 17.
El aceite de Lorenzo, una mezcla de gliceroltrioleato y gliceroltrierucato, podría normalizar los niveles plasmáticos de AGCML y en teoría retrasar la progresión de formas cerebrales leves de la enfermedad y en la adrenomieloneuropatía; sin embargo, los resultados de los estudios actuales con respecto a los beneficios clínicos aún son conflictivos y, puntualmente en adrenomieloneuropatía, la información es escasa. Soportado por varios estudios observacionales, el trasplante de células madre hematopoyéticas se considera una buena opción de tratamiento en niños varones con compromiso cerebral en fases tempranas, aunque esta práctica no ha sido adecuadamente estudiada en adrenomieloneuropatía 18,19.
CONCLUSIONES
La adrenoleucodistrofia, si bien es una entidad rara, debe ser considerada dentro de los diagnósticos diferenciales en el contexto de pacientes adultos con síndromes neurológicos crónicos progresivos o recurrentes, principalmente mielopatías sin causa clara y en especial cuando existe asociación con insuficiencia suprarrenal o existe algún antecedente familiar. Las posibilidades terapéuticas hoy en día son escasas, y la evidencia con los tratamientos específicos disponibles en adultos es insuficiente. Sin embargo, la posibilidad de tener un diagnóstico traería beneficios, evitando estudios innecesarios, alejando la incertidumbre diagnóstica por parte del médico y del paciente y determinando un pronóstico de la enfermedad a mediano y largo plazo.

