











 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
Introduction
Peutz-Jeghers syndrome (PJS) is a rare, autosomal dominant, hereditary polyposis associated with a high frequency of STK11/LKB1 gene mutations located on the short arm of chromosome 191,4. It is characterized by mucocutaneous pigmentation and multiple hamartomatous polyps, with an increased risk of gastrointestinal malignancy. It has also been associated with an elevated risk of gynecological, testicular, and thyroid cancer1.
Mucocutaneous pigmentation is due to melanin-laden macrophages in the dermis and increased melanocytes at the dermal-epidermal junction, present in about 95% of patients. These usually appear in childhood around the mouth, nostrils, perianal region, fingers, and knuckles2,5.
PJS polyps are usually located in the small intestine, mainly in the jejunum, followed by the colon, rectum, and stomach. They can also appear outside the gastrointestinal tract, including the renal pelvis, bladder, lungs, and nasopharynx5. Histological features include an elongated wave-shaped epithelial component, a cystic dilatation of the gland extending into the submucosa, and smooth muscle extending into the fronds of polyps2.
The clinical diagnosis should be considered in an individual meeting at least one of the following criteria1-3:
Clinical case
We present the case of a 25-year-old man from the urban area of the municipality of Barbosa (Santander), who was admitted to the emergency department due to diffuse abdominal pain of three years of evolution and rectal bleeding. He had a history of right hemicolectomy for acute abdomen secondary to ileocolonic intussusception due to a giant polyp in the terminal ileum measuring 45 cm x 42 cm. Upon checking the surgical specimen, another giant polyp was found in the jejunum, 80 cm from the angle of Treitz, both with hamartomatous histopathology. The physical examination on admission revealed multiple hyperchromatic dark brown macular lesions on the buccal mucosa, with no other relevant findings (Figure 1).

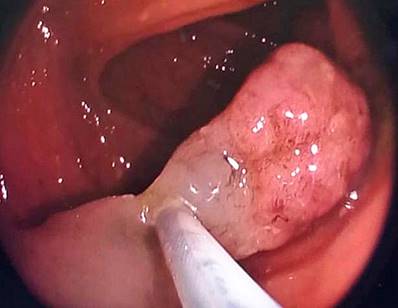
The patient was hospitalized and underwent a colonoscopy, noting two 7 mm polyps near the rectum and two pedunculated polyps in the proximal transverse muscle measuring 20 mm x 15 mm (Figure 2) and 20 mm x 20 mm (Figure 3), respectively. An upper digestive endoscopy was also performed, documenting multiple sessile polyps (around ten) between the first and second duodenal portions, with diameters ranging from 4 mm to 12 mm (Figures 4 and 5). All colon polyps were endoscopically resected, and one of the largest duodenal polyps was removed.



The histopathological report of the colon polyps was of the hamartomatous type, and one was a tubular adenoma without dysplasia (Figures 6 and 7). The resected duodenal polyp was reported as a hamartomatous polyp.
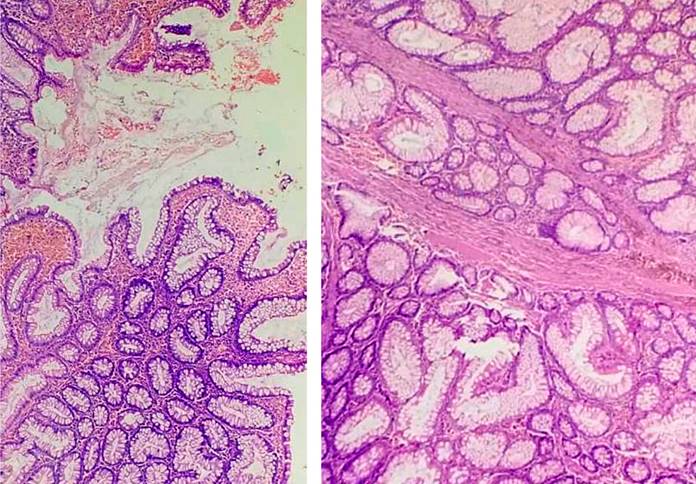
Figure 6 Polypoid lesions made up of glands of various shapes and sizes, supported by bands of smooth muscle fibers. Source: Authors’ archive.

Discussion
Peutz-Jeghers syndrome is a rare, autosomal dominant disease with few reports in our country. According to Wu et al., it has an estimated incidence of one case per 25,000-300,000 live births. It may occur in any ethnic group and affects men and women alike. Its average age of diagnosis is 23 years, and the average age for cancer development is 42 years6.
Moreover, according to Beggs et al., the first manifestation is often gastrointestinal obstruction due to intussusception by polyps, 69% of the time in the small intestine, and usually occurs between 6 and 18 years of age2,6.
The main goal in effectively treating patients with Peutz-Jeghers syndrome is based on surveillance, prevention, and treatment of complications. Recommendations include6-8:
○ It is initially detected from the age of 12.
○ If polyps are found, repeat annually.
○ Repeat every 2-3 years until adulthood in the absence of polyps
○ Initial screening starts at age 12 or earlier if symptoms are reported.
○ If polyps are found, repeat annually.
○ In the absence of polyps, repeat at intervals of 1-3 years
○ Magnetic resonance cholangiopancreatography (MRCP), biliopancreatic endoscopic ultrasound, or both: from 25-30 years of age; repeat every 1-2 years
○ Breast exam: clinical examination every six months from the age of 25.
○ Mammography: from the age of 25
Finally, in terms of prevention, we advise providing genetic counseling to couples with a family history of Peutz-Jeghers syndrome who wish to have children6-10.
Conclusions
Peutz-Jeghers syndrome is a rare disease but with relevant clinical connotations. These patients have a 15-fold increased risk of gastrointestinal malignancy, although it has also been associated with extraintestinal neoplasms.
It is crucial to know its initial clinical manifestation: hyperpigmented mucocutaneous macules (present in more than 95% of cases), a positive family history of Peutz-Jeghers syndrome, and hamartomatous-type gastrointestinal polyposis.
Thus, a prompt diagnosis would be achieved to implement adequate surveillance, as previously recommended, to avoid complications such as intestinal obstruction secondary to intussusception and emergency surgical interventions and monitor the appearance of premalignant and malignant lesions.

