











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
El linfoma de Hodgkin (LH) es un proceso linfoproliferativo clonal originado en células B del centro germinal. Compromete predominantemente cadenas de ganglios linfáticos cervicales, mediastinales y axilares, y su historia natural se caracteriza por el crecimiento tumoral lento y sostenido, inicialmente en un estadio localizado, extiendiéndose en forma progresiva a lo largo de diferentes cadenas ganglionares y sitios extraganglionares. Para la estadificación de la enfermedad al momento del diagnóstico se emplea la clasificación de Ann Arbor con la modificación de Cotswolds, que se basa en la extensión anatómica de la enfermedad, asociado o no a la presencia de síntomas B y/o enfermedad voluminosa (masa bulky - ensanchamiento mediastinal mayor a 10 cm o más de 1/3), determinándose cuatro estadios, definiéndose enfermedad localizada a los Estadios I y II (no bulky) y enfermedad avanzada a los Estadios II (bulky) III y IV1,2.
Afecta a todos los grupos etarios, con un pico bimodal en adultos jóvenes y en adultos, con una tasa de incidencia mundial ajustada por edad para 2012 de 0,9 casos cada 100.000 individuos, siendo mayor en países más desarrollados (2,1 casos cada 100.000) que en países menos desarrollados (0,65 casos cada 100.000)3. La misma tasa en América Latina (2012) es de 1 caso cada 100.000 individuos3, observándose las mayores incidencias en Argentina 2003-2007 (1,8/100.00), Brasil 2003-2007 (1,8/100.00), Uruguay 2005-2007 (1,7/100.00), Costa Rica 2003-2007 (1,6/100.00) y Cuba 2004-2007, (1,7/100.00) y las menores incidencias en Bolivia 2011 (0,5/100.00) y El Salvador 1999-2003 (0,4/100.00)4. El LH representa estimativamente el 10% del total de los linfomas diagnosticados, ascendiendo al 21% cuando se analiza su frecuencia dentro de los linfomas en edad adolescente5. Presenta una tasa de supervivencia aproximada de 92% al año, 85% a los 5 años y 80% a los 10 años del diagnóstico1.
Desde el punto de vista histológico, se caracteriza por presentar escasas células neoplásicas, denominadas células de Reed Sternberg/Hodgkin (RS/H) y sus variantes citomorfológicas (1-2% de la masa tumoral), las cuales se encuentran inmersas en un profuso microambiente tumoral, rico en células inflamatorias pertenecientes al sistema inmune (linfocitos, histiocitos, plasmocitos, mastocitos, neutrófilos y eosinófilos). Actualmente, el método diagnóstico de elección es el estudio histomorfológico mediante biopsia de la lesión tumoral, acoplado a su caracterización inmunofenotípica mediante técnicas de inmunohistoquímica, cuyos criterios diagnóstico mínimos se basan en la identificación de las células de RS/H y/o sus variantes, en un contexto inflamatorio determinado, demostrando un perfil inmunofenotípico característico6. De este modo, se logran identificar dos entidades clínico-patológicas bien diferenciadas dentro del LH, el LH clásico (LHC), el cual comprende el 95% de los casos de LH y el LH predominio linfocítico nodular. A su vez el LHC se subdivide en cuatro subtipos histológicos: esclerosis nodular, celularidad mixta, rico en linfocitos y depleción linfocitaria, basándose en diferentes características de la composición y estructura del microambiente tumoral6, 7.
Si bien la descripción clínico-patológica del LH se remonta a la primera mitad del siglo XIX8, hace casi 200 años, en la actualidad el LH sigue siendo una patología que despierta mucho interés científico por múltiples factores, como su heterogeneidad clínica, su amplio rango etario de aparición, su complejidad histológica, la variabilidad en la expresión de marcadores de superficie, la particular desregulación de múltiples vías de señalización y los resultados terapéuticos favorables alcanzados hasta la actualidad9,10,11,12.
Aunque no resulta claro el rol exacto del infiltrado celular inflamatorio/inmunitario presente en el LH, hay evidencia que indica que las células de RS/H desarrollan diversas estrategias tendientes a atraer diferentes tipos de células inflamatorias/inmunes, que disminuyen la actividad citotóxica del sistema inmune antitumoral, promoviendo un microambiente pro-tumoral acorde a las necesidades de supervivencia de las células neoplásicas. A su vez, las mismas células del sistema inmune producen citosinas y quimiosinas que favorecen la proliferación de las células de RS/H, promoviendo su expansión clonal13,14. De esta forma las células de RS/H desarrollan la capacidad necesaria para la formación y perpetuación de un entorno que promueve el crecimiento tumoral, mientras que a su vez bloquea los mecanismos inflamatorios fisiopatológicos antitumorales, favoreciendo al escape inmunitario.
Diferentes estudios relacionados al microambiente tumoral en LHC, han descripto múltiples mecanismos celulares y moleculares interrelacionados, que permiten colectivamente a las células de RS/H resistir al ataque de las defensas innatas y adaptativas del sistema inmune del huésped, entre los cuales se incluyen: mutaciones inactivantes del antígeno leucocitario humano, sobreexpresión de moléculas de membrana celular inmunotolerogénicas, creación de una red de citocinas inmunosupresoras, reclutamiento de células inmunes inmunosupresoras y activación de la vía coinhibitoria PD-1/PD-L1-2, entre otras15.
A nivel genético, las células de RS/H se caracterizan por presentar un gran número de aberraciones cromosómicas complejas, relativamente recurrentes, lo que refleja su marcada inestabilidad genómica16,17,18. Por otro lado, existe escasa evidencia de alteraciones cromosómicas en células del microambiente tumoral19,20. En este contexto, datos recientes de la literatura, describen que la amplificación del locus 9p24.1 en células de RS/H es una anomalía altamente recurrente, tanto en líneas celulares como en tumores primarios21,22. Esta región cromosómica resulta de importancia, dado que en ella mapean genes involucrados en la modulación de mecanismos de inmunotolerancia, como PD-L1 (Programmed death 1 Ligand 1) y PD-L-2 (Programmed death 1 Ligand 2) así como también el gen JAK2 (Janus Kinasa 2), relacionado a la vía de señalización JAK/STAT (Janus kinasa/Signal transducer and activator of transcription proteins)23 y el gen JMJD2C (Jumonji domain-containing protein 2C), el cual codifica para una histona demetilasa, pudiendo jugar un rol importante en la remodelación epigenética de la célula RS/H24.
Desde el punto de vista del análisis mutacional, los genes alterados con mayor recurrencia se vinculan a la vía de señalización de NF-kB (Nuclear Fcator Kappa B) y JAK/STAT, las cuales están constitutivamente activadas en las células de RS/H. Estas mutaciones no son mutuamente excluyentes, observándose múltiples genes alterados para una misma vía. Las mutaciones vinculadas a la vía de NF-kB se producen predominantemente en inhibidores de la señalización, como TNFAIP3 (TNF Alpha Induced Protein 3) (~40%)25, NFKBIA (~15%)26 y NFKBIB (~15%)27 que, en conjunto con alteraciones numéricas y estructurales de genes activantes relacionados, como la amplificación de REL (REL Proto-Oncogene, NF-KB Subunit), la amplificación de MAP3K14 (Mitogen-Activated Protein Kinase Kinase Kinase 14) y la translocación o ganancia de BCL3 (B cell leukemia/lymphoma 3), potencian aún más la activación constitutiva de esta vía. Con respecto a la vía de señalización de JAK/STAT, además de observarse amplificación del gen JAK2 como ya se mencionó, se reportan mutaciones inactivantes en el regulador negativo SOCS1(Suppressor Of Cytokine Signaling 1) (~40%) y mutaciones activantes en STAT6 (~30%)28.
Particularmente, PD-L1 y PD-L2 son proteínas de transmembrana tipo 1 que se encuentran codificadas por los genes CD274 (CD274 Molecule) y PDCD1LG2 (Programmed Cell Death 1 Ligand 2), respectivamente. Ambas están constitutivamente expresadas en diferentes tipos de células: macrófagos, células presentadoras de antígenos, endoteliales, sincitiotrofoblasto placentario y pulmón, entre otras, las cuales participan de la homeostasis del sistema inmune mediante la vía de señalización PD-1/PD-L1-229.
Por su parte, PD-1 (Programmed death 1) es una molécula de señalización de la superficie celular que se expresa en linfocitos T activados, así como en otras células efectoras del sistema inmune: NK, células B, macrófagos, linfocitos T reguladores y células T foliculares. En condiciones fisiológicas y mediante la interacción con sus ligandos, PD-L1 y PD-L2, envían una señal inhibitoria que regula el balance entre la activación de células T y los mecanismos biológicos de tolerancia inmunológica, lo que permite controlar la intensidad y duración de la respuesta inmune, limitando el daño tisular en respuesta a determinado antígeno, y el posible desarrollo de enfermedades autoinmunes y alérgicas30.
En consecuencia, la estimulación de la vía de señalización PD-1/PD-L1-2 genera un fenómeno de agotamiento de las células T efectoras (células T “exhaustas”), resultando en la inhibición temporaria y reversible de su activación, así como limitando su diferenciación y expansión clonal21,29,30. Además de su importancia en un contexto fisiológico como el ya mencionado, este mecanismo de inmunomodulación es de sumo interés debido a que es utilizado como herramienta biológica por diversos tipos de neoplasias, para escapar a los mecanismos de vigilancia antitumoral del sistema inmune, favoreciendo una respuesta antigénica crónica inefectiva y asimismo la persistencia y/o progresión de la enfermedad clonal. Diferentes estudios han reportado altos niveles de expresión de PD-L1 en tumores sólidos31,32,33,34 y neoplasias hematológicas, como LH, linfoma de células B primario mediastinal, linfomas de células T y linfoma difuso de células grandes B primario del sistema nervioso central y testicular21,35,36,37.
En este contexto, el objetivo del presente trabajo es caracterizar los desbalances genómicos del locus PDL1/PDL2 ubicado a nivel de 9p24.1 en biopsias de pacientes fijadas en formol y embebidas en parafina con diagnóstico histopatológico e inmunofenotípico de LHC, tanto en las células RS/H como en las células del microambiente tumoral, y correlacionarlo con el nivel de expresión proteica de PD-L1, la presentación de la enfermedad, el estadio tumoral según Ann Harbor y los resultados de la evaluación interina por tomografía por emisión de positrones a los 3 meses postquimioterapia (PET3).
Materiales y Métodos
Se evaluaron, en forma retrospectiva y secuencial biopsias completas de 22 pacientes con diagnóstico histológico e inmunofenotípico de LHC, fijadas en formol y embebidas en parafina, correspondientes al período 2011-2014. El estudio fue aprobado por el Comité de Ética de los Institutos de la Academia Nacional de Medicina. Todos los pacientes dieron su consentimiento informado. Como criterios de inclusión, se estableció trabajar con biopsias correspondientes al diagnóstico inicial de la enfermedad, de pacientes mayores de 18 años, con serología negativa para el virus de la inmunodeficiencia humana y sin tratamiento previo de la enfermedad. Los parámetros clínicos analizados fueron obtenidos del registro de pacientes que formaron parte del estudio GATLA LH-05 realizado en la República Argentina2.
Para evaluar desbalances genómicos a nivel del locus 9p24 se efectuó el análisis por FISH (Fluorescence in situ hybridization) del clúster PD-L1/PD-L2 empleando la sonda SPEC CD274/PDCD1LG2/CEN9 (ZytoVision, Alemania), acorde a las indicaciones del fabricante, y a lo previamente descripto38. Se determinó el número de copias del locus PD-L1/PD-L2 en relación al centrómero del cromosoma 9 analizando 50 células de RS/H para cada caso, clasificando las alteraciones genómicas como amplificación (A) cuando había ≥3 copias del gen en estudio respecto al número de señales centroméricas (ratio ≥3:1), ganancia cuando el ratio era <3:1 pero >1:1 y polisomía cuando el ratio era 1:1 (Figura 1). Además, se evaluaron 3 biopsias de linfadenitis reactivas (tejido linfoide reactivo), como controles para la detección de monosomía del cromosoma 9 en células del microambiente tumoral, determinando como punto de corte 20%, calculado como la media +3 desvíos estándar. Por último, se efectuó inmunomarcación sobre cortes completos de las mismas biopsias con el anticuerpo anti-PD-L1/CD274 (clon RBT-PDL1, Rmab, BioSB) para analizar el nivel de expresión proteica en células de RS/H, considerando grado +3 el máximo nivel (tinción de intensidad moderada/fuerte >30% de las células tumorales), seguido por grado +2 (tinción de intensidad moderada/fuerte en el 5 a 30% de las células tumorales), +1 (tinción de intensidad débil >5%) y 0 (con ausencia de expresión o expresión <5%) (Figura 2), adaptado de las técnicas de evaluación de estudios previos en neoplasias linfoides39,40,41.
Para el análisis estadístico se empleó el programa IBM SPSS Statistics 24. La comparación de los datos de los pacientes se efectuó utilizando el test de Mann-Whitney. El análisis de las variables cuantitativas se realizó empleando el test “t” de Student en tanto que para las variables categóricas se utilizó la prueba de Chi cuadrado o el Test Exacto de Fisher. Se consideró significativo un p<0,05.

Fuente: autores
Figura 1 Células de Reed Sternberg/Hodgkin de pacientes con linfoma de Hodgkin clásico hibridadas con la sonda SPEC CD274/PDCD1LG2/CEN9 (locus 9p24.1: verde, centrómero del cromosoma 9: rojo) (ZytoVision, Alemania) mostrando: A) Amplificación del locus 9p24.1; B) Ganancia; C) Polisomía.

Resultados
Los pacientes presentaron una edad media de 31 años (rango 18 a 53 años), siendo el 50% del sexo masculino. Al momento del diagnóstico el 41% (9/22) tenía un estadio temprano de la enfermedad (I y II no bulky) y el 59% (13/22) un estadio avanzado (II bulky, y IV). El 36% (8/22) de los casos desarrolló síntomas B, mientras que el 27% (6/22) de los pacientes presentó masa bulky al diagnóstico, representando el 46% (6/13) de los casos en estadio avanzado (Tabla 1). Todos pertenecían al subtipo histológico de LHC esclerosis nodular.
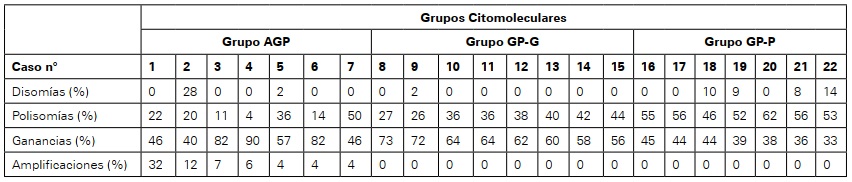
El análisis por FISH de las células de RS/H mostró alteraciones en el número de copias del locus PD-L1/ PD -L2 en la totalidad de los casos, identificándose un espectro de alteraciones numéricas con porcentajes variables de A, G y P, para cada caso (Tabla 2). Esto permitió diferenciar dos grupos citomoleculares: uno con presencia de los tres tipos de alteraciones (Grupo AGP) (7; 32%) y otro sin amplificación (Grupo GP) (15; 68%). A su vez, dentro del grupo GP se observó un 53% de casos (8/15) con >50% de células de RS/H con G, (GP-G; GP rico en ganancias), en tanto que el 47% (7/15) restante presentó mayor porcentaje de polisomías (GP-P; GP rico polisomías). No se encontró ningún paciente que presentara un solo tipo de alteración.
Resulta interesante destacar que los pacientes del grupo AGP mostraron menor edad media al inicio de la enfermedad (26 años) que el grupo GP (34 años) (Tabla 1). Cuando este último grupo lo dividimos en los dos subgrupos previamente mencionados, vemos que el grupo GP-G presentaba una media de edad menor (30,7 años) que el GP-P (37,7 años). No se observaron diferencias significativas entre AGP y GP-G por lo que fueron analizados en conjunto, mostrando una edad media significativamente menor (28,6 años) que la del grupo GP-P (p=0,027). Asimismo, el análisis de las características clínicas mostró ausencia de masa bulky en los pacientes del grupo GP-P, manifestación que si fue observada en los casos AGP y en aquellos con GP-G (6/15; 40%), indicando un comportamiento más agresivo de la enfermedad. No se observaron diferencias significativas en relación al estadio de la enfermedad y la presencia de síntomas B (Tabla 1). Por último, el análisis del PET fue positivo (Deauville score 3-4-5)42 en el 43% (3/7) de los casos con amplificación, respecto del 27% (4/15) de los pacientes sin amplificación.
Tabla 1 Características clínico – patológicas de los pacientes con LHC estudiadosF: femenino; M: masculino; A: amplificación; G: ganancia: P: polisomía

Fuente: autores
Tabla 2 Espectro de alteraciones numéricas del locus 9p24.1 en células de RS/HGrupo AGP: grupo con amplificaciones, ganancias y polisomías); Grupo GP-G: grupo con ganancias y polisomías, rico en ganancias (G>50%); Grupo GP-P: grupo con ganancias y polisomías, rico en polisomías (P>50%)
Fuente: autores
A nivel proteico, se detectó expresión de intensidad variable para PD-L1 en células de RS/H en el 95% de los casos (21/22). Cuando efectuamos el análisis por grupo, se observó que los pacientes del grupo GP-P tenían menor nivel de expresión, con score +1 en el 57% de los pacientes (4/7), en tanto que el score +3 se visualizó solamente en el grupo AGP (4/7; 57%) y en un caso del subgrupo GP-G (1/8; 12,5%). No se observaron casos con mínima o nula expresión en el grupo AGP, indicando una asociación positiva entre el número de copias del locus PD-L1/PD-L2 y el nivel de expresión proteica.
Por otra parte, el análisis de los linfocitos pequeños del microambiente tumoral circundantes a las células RS/H mostró monosomía del cromosoma 9 en el 53% de los casos (Figura 3a). Esta alteración se encontró significativamente asociada al grupo GP-P (100% de los casos) respecto del grupo AGP y GP-G (36,4%) (Figura 3b), sugiriendo una mayor inestabilidad genómica en las células del microambiente tumoral en este subgrupo.
Discusión
En el presente trabajo efectuamos el análisis de los desbalances genómicos del locus 9p24.1 mediante FISH tanto en las células de RS/H como en las del microambiente tumoral, así como la evaluación de la expresión proteica de PD-L1, en material de biopsias incluidas en parafina de pacientes con LHC. Los resultados fueron comparados con algunas características clínicas de la patología, como la edad, el estadio tumoral y la respuesta al PET3.

Fuente: autores
Figura 3 Análisis de los linfocitos pequeños del microambiente tumoral circundantes a las células Reed Sternberg/Hodgkin (RS/H). A)Núcleosinterfásicos hibridados con la sonda SPEC CD274/PDCD1LG2/CEN9 (locus 9p24.1: verde, Centrómero del cromosoma 9: rojo) (ZytoVision,Alemania) mostrando: célula de RS/H (flecha roja), linfocitos circundantes disómicos (flechas amarillas), linfocitos con monosomía del cromosoma 9 (punta de flecha blanca). B) Gráfico mostrando la distribución de la monosomía del cromosoma 9 en los distintos subgrupos citomoleculares. Cr9: cromosoma 9; A: amplificación; G: ganancia; P: polisomía.
El análisis de nuestros datos mostró desbalances genómicos, fundamentalmente ganancia del número de copias y amplificación del locus 9p24.1, en las células de RS/H de todos los casos estudiados. Este valor resulta similar al detectado por Roemer et al (92%), en donde el 97% de la población estudiada presentaba alteraciones numéricas de dicho locus. Por el contrario, contrasta con los resultados de estudios realizados empleando CGH (Comparative genomic hybridization) en células de RS/H aisladas por microdisección láser, que mostraron mucho menor frecuencia de aumento del número de copias para dicho locus (40%)16,17.
Nuestra serie mostró amplificación del locus 9p24.1 en el 32% de los casos, sustentando datos previos de la literatura que detectan esta alteración en el 36-38% de los pacientes21,22. Por el contrario, nuestra cohorte no presentó casos sin alteraciones numéricas del locus 9p24.1 o con polisomías en forma exclusiva, los cuales sí fueron detectados en el trabajo de Roemer et al22, aunque en muy baja frecuencia (1% y 5%, respectivamente).
Asimismo, resulta interesante destacar que, en nuestra serie, los pacientes con amplificación y los del subgrupo GP-G presentaron una edad media significativamente menor que los pertenecientes al subgrupo GP-P. Por otro lado, los grupos AGP y GP-G mostraron asociación con la presencia de masa bulky, relacionada a enfermedad progresiva, aspecto no detectado en los pacientes GP-P. Esta observación resulta de particular importancia, sustentando un probable valor pronóstico de la amplificación de 9p24.1 en LHC. No obstante, la literatura muestra datos contradictorios. Roemer et al22 observaron una menor sobrevida libre de progresión (SLP) y estadios más avanzados al debut de la enfermedad en los casos con amplificación del locus PD-L1/PD-L2, sugiriendo que la evasión de este mecanismo biológico del sistema inmune antitumoral por parte de las células de RS/H, disminuye la capacidad de contención del clon neoplásico en su sitio anatómico, favoreciendo la progresión de la enfermedad. En el mismo sentido, Paydas et al41 describieron peor SLP y sobrevida global (SG) en casos de LHC con co-expresión de PD-L1 y PD-1 en células tumorales y del microambiente tumoral. Por el contrario, otros autores no encuentran diferencias en la SLP y SG entre los casos con alta y baja expresión de PD-L1/PD-L2 en células de RS/H43,44,45. Por su parte, Hollander et al46 observan diferencias en la SLP en los casos con alta expresión de PD-L1 y PD-1 en células del microambiente tumoral y no en células de RS/H, siendo necesario continuar estos estudios a fin de definir el significado clínico de esta alteración en el LHC.
Simultáneamente, nuestro análisis también mostró asociación positiva entre el número de copias del locus PD-L1/PD-L2 con el nivel de expresión proteica de PD-L1 por inmunohistoquímica en células de RS/H. Esta asociación fue demostrada inicialmente por Green et al21 quienes también observaron el aumento de expresión de JAK2. Cabe destacar que el amplicón 9p24.1 incluye los genes CD274 y PDCD1LG2 y la secuencia codificante para JAK2, una tirosina kinasa que media la activación de receptores de citosinas involucrados en mecanismos de inflamación e inmunidad, acoplada a la vía de señalización JAK-STAT29. Esto resulta de importancia ya que la amplificación del locus 9p24.1 no solo implica modificaciones en el número de copias de PD-L1 y PD-L2, sino también del gen JAK2, lo que conduce a una mayor expresión proteica y la subsecuente activación de la vía de señalización JAK/STAT, induciendo aún más la expresión de los ligandos de PD-1, profundizando la activación del eje PD-1/PD-L1/2, lo que en definitiva lleva a mayor proliferación neoplásica21,35. En este contexto, recientemente Carey et al46 realizaron un estudio topográfico de las células del microambiente tumoral del LHC mediante inmunofluorescencia multiplexada y análisis digital de imágenes, concluyendo que los macrófagos asociados a tumor son las células del microambiente tumoral que más expresan PDL-1 y se co-localizan espacialmente en las inmediaciones de las células de RS/H PD-L1+, fortaleciendo sinérgicamente la activación de la vía de señalización PD1-PD-L1/2 de los linfocitos T efectores, disminuyendo la capacidad de reacción del sistema inmune antitumoral.
Por otra parte, diferentes estudios mostraron la presencia de aberraciones cromosómicas, particularmente alteraciones numéricas, en células linfoides morfológicamente normales del microambiente tumoral, localizadas en la proximidad de las células neoplásicas19,20. En nuestra serie, observamos monosomía del cromosoma 9 en las células del microambiente tumoral circundantes a las células RS/H en el 53% de los casos, particularmente asociado al grupo GP-P. Este hallazgo sustentaría la inestabilidad cromosómica descripta en pacientes con LH, manifestada por la presencia de rearreglos cromosómicos complejos en las células de RS/H y aneuploidías tanto en las células neoplásicas como en los linfocitos aparentemente normales del microambiente tumoral35,47,48, así como también por estudios en linfocitos de sangre periférica de pacientes con LHC pre y post-tratamiento49,50,51y análisis de micronúcleos y disfunción telomérica18. El empleo de metodologías más recientes como CGH y NGS (Next generation sequencing) también detectan una alta inestabilidad cromosómica en estos pacientes16,17, 51
Conclusiones
Si bien nuestro estudio presenta limitaciones relacionadas con su carácter retrospectivo y el bajo número de pacientes evaluados, los resultados obtenidos muestran que los desbalances genómicos del locus PD-L1/PD-L2 son altamente frecuentes en el LHC en nuestra región, presentándose con un espectro de alteraciones numéricas que permitió diferenciar claramente dos grupos distintivos. Los casos con amplificación de 9p24.1 mostraron una menor edad de inicio de la enfermedad, masa bulky mediastinal al diagnóstico, aumento significativo de la expresión proteica de PD-L1 y menor inestabilidad genómica en las células del microambiente tumoral. Sin duda, estos hallazgos constituyen un aporte a la caracterización biológica del LHC, siendo de interés en el marco de las nuevas modalidades terapéuticas con inmunomoduladores11,12 que demostraron que el bloqueo de esta vía de señalización inhibitoria de linfocitos T efectores, se asocia a una mejor tasa de respuesta al tratamiento y de SLP, alcanzando una alta eficacia terapéutica.

