











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
La dermatosis pustular subcórnea (DPS), también conocida como enfermedad de Sneddon-Wilkinson es una erupción pustulosa estéril infrecuente (1) que hace parte de las dermatosis neutrofílicas (2-4). Afecta principalmente a las mujeres, en una proporción de 4:1 con respecto a los hombres. Se suele presentar entre la quinta y séptima décadas de vida (4-6), aunque también se han descrito casos en niños (3,5).
Se trata de una enfermedad crónica y recidivante, caracterizada por la aparición de pústulas flácidas que se diseminan rápidamente (4,6). La erupción es simétrica y se localiza principalmente en el tronco, las extremidades proximales, las superficies flexoras y las áreas intertriginosas (1-6). Es una enfermedad difícil de reconocer debido a su amplio espectro de diagnósticos diferenciales (2,4-6). A continuación, se describe la experiencia de los autores en el diagnóstico y tratamiento de seis pacientes colombianos con esta enfermedad.
Reporte de casos
Se presenta una serie de casos de seis mujeres con DPS, con edades entre 45 y 82 años (Tabla 1), diagnosticadas en un centro dermatológico de referencia nacional entre el 2010 y el 2021. Las pacientes presentaban lesiones de larga evolución, en promedio de cinco años, con predominio de pústulas, pápulas y placas eritematocostrosas con bordes circinados, además de vesículas, ampollas, erosiones y costras (Figura 1). Las zonas más afectadas fueron el tórax, las áreas intertriginosas y el tercio proximal de las extremidades.
Tabla 1: Características sociodemográficas, clínicas e histológicas de las seis pacientes de la serie
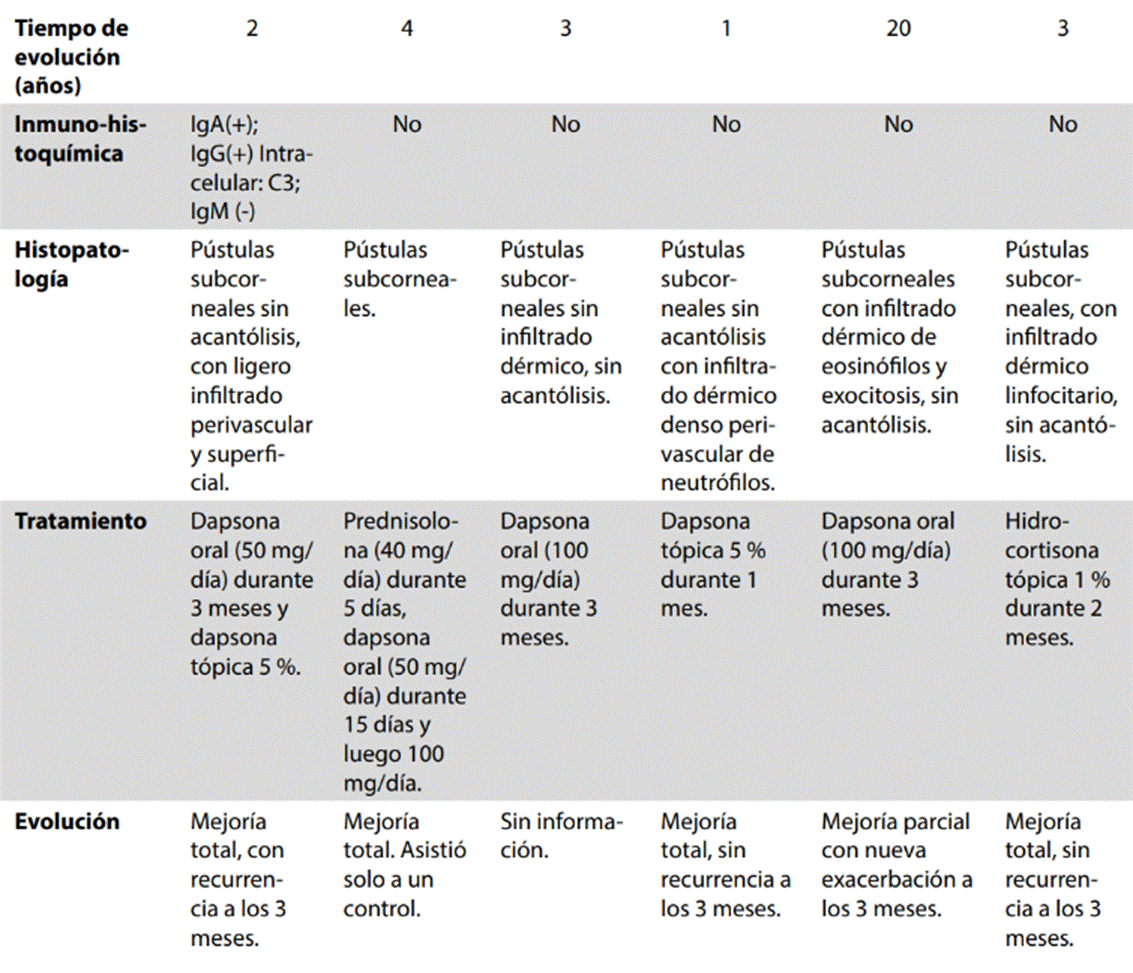
Fuente: creación propia
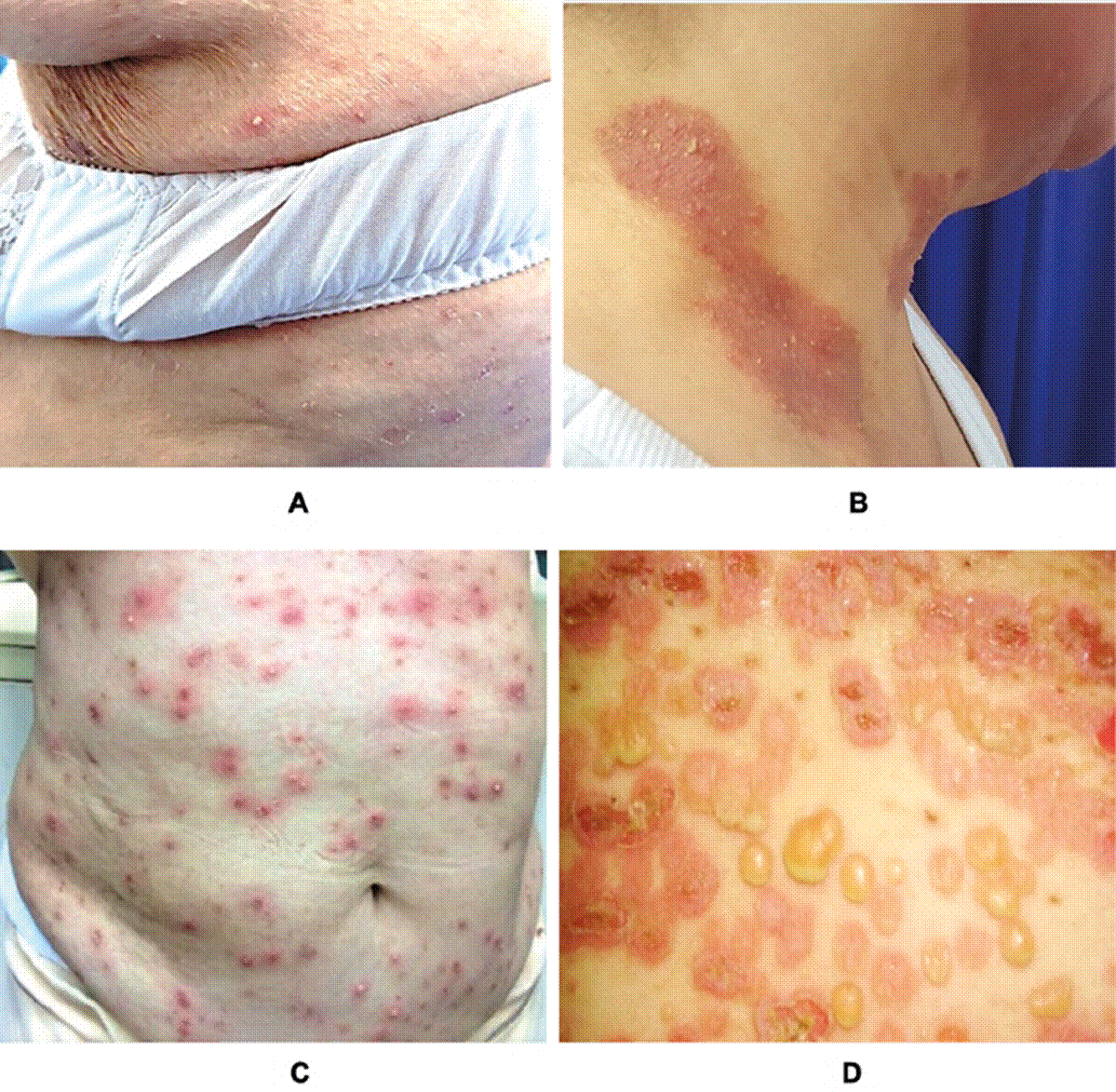
Figura 1: Lesiones clínicas de pacientes con dermatosis pustular subcórneal. A. Pústulas en la región axilar, pápulas y placas erosivas en el tórax. B. Placa eritematocostrosa con bordes descamativos en el cuello. C. Pápulas y pústulas de base eritematosa en el tórax y el abdomen. D. Ampollas y pústulas hipopión, algunas erosionadas y cubiertas por costras. Fuente: creación propia
En dos casos se observaron las pústulas flácidas características de DPS, con material purulento en su base y contenido seroso en la parte superior (Figura 2). Todas las pacientes presentaron prurito, con excepción del caso número seis.
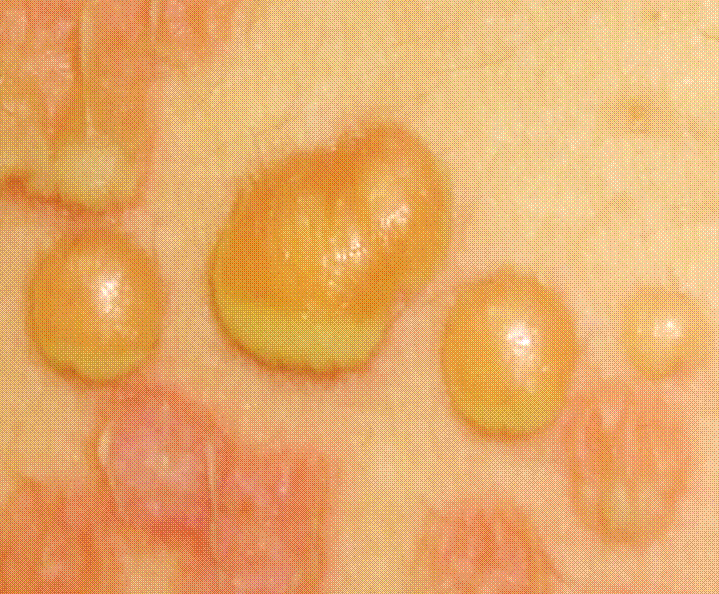
Figura 2: Acercamiento de las pústulas hipopión características de la DPS. Se observa el contenido purulento en la base de la lesión, que contrasta con el líquido seroso de la parte superior. Fuente: creación propia
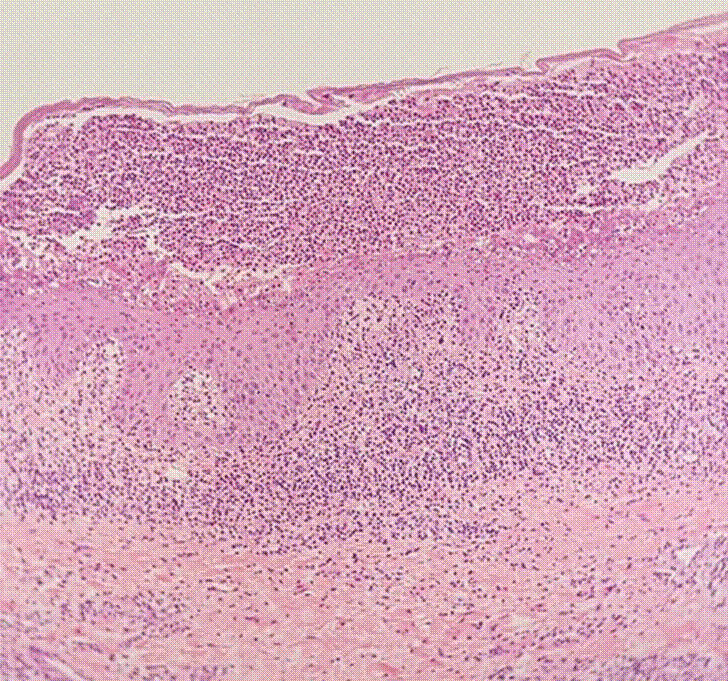
Todos los casos cumplieron los criterios clínicos descritos en la literatura (Tablas 2 y 3) y se analizó la correlación con los hallazgos identificados en la histopatología: acumulación subcorneal de neutrófilos en ausencia de espongiosis y acantólisis (Figura 3). Se realizó inmunofluorescencia directa (IFD) solo en el primer caso, con presencia de anticuerpos IgA e IgG positivos a nivel intercelular, C3 e IgM negativos (Tabla 1).
Discusión
La clasificación de la DPS ha sido controversial desde su primera descripción. En 1956, Sneddon y Wilkinson la diferenciaron de la dermatitis herpetiforme y el pénfigo (1), y establecieron sus características clínicas e histológicas (Tabla 2).
Tabla 2: Principales características de los pacientes de la primera serie de casos de DPS reportada en 1956 (1)

Fuente: creación propia
Posteriormente, otros autores cuestionaron que la DPS fuera considerada una enfermedad independiente, y argumentaron que los hallazgos descritos por Sneddon y Wilkinson eran inespecíficos y se trataba de una variante de psoriasis pustulosa (7,8). En cuanto a la histopatología, en la psoriasis pustulosa es usual encontrar hiperplasia psoriasiforme, paraqueratosis y pústulas espongiformes (9), hallazgos no comunes en la DPS.
En la actualidad, la DPS se incluye dentro de las dermatosis neutrofílicas (2,3), aunque la literatura sobre el tema es limitada. Su incidencia es desconocida, con pocos casos reportados en Latinoamérica (9-11). Esta serie reúne todos los pacientes con diagnóstico confirmado de DPS en un centro de referencia en dermatología durante un periodo de 11 años.
El daño tisular que ocurre en la DPS es secundario a un proceso inflamatorio de origen incierto, derivado de la migración de neutrófilos hacia la parte superior de la epidermis (5). La hiperactivación de los neutrófilos bajo el estrato córneo es favorecida por niveles elevados de factores quimiotácticos, como interleucina (IL) 1β, IL-6, IL-8, IL-10, leucotrieno B4, complemento C5a, IgA y factor de necrosis tumoral alfa (TNF-α) (4,12). Es probable que las variaciones que se presentan en este perfil de mediadores proinflamatorios se relacionen con el espectro de manifestaciones clínicas, severidad y respuesta al tratamiento (13).
En cinco pacientes de la serie (83 %) se describieron pústulas, además de otras lesiones; sin embargo, solo en dos (33 %) se documentó la pústula hipopión. Estas pústulas pueden ser difíciles de identificar, pues su rápida y espontánea ruptura conduce a la formación de erosiones y placas escamocostrosas (4). El aspecto macroscópico de la pústula, con apariencia alargada o flácida (se compara con los nidos colgantes de las aves oropéndolas), explica el uso del término hipopión, que ocurre en algunas uveítis por acumulación de leucocitos y fibrina en la cámara anterior del ojo (14,15). Sin embargo, la enfermedad no se suele sospechar cuando predominan las erosiones o las placas escamocostrosas (ver Figura 2B).
En 1981, Pinkus definió seis criterios diagnósticos (16) que complementan la descripción original de Sneddon y Wilkinson (Tabla 3).

Teniendo en cuenta lo anterior, la sospecha clínica de DPS no debe partir exclusivamente de la presencia de una pústula hipopión. Cualquier erupción vésicopustulosa crónica en mujeres de mediana edad con afectación de zonas anatómicas características demanda una historia clínica completa, con el objetivo de obtener la información necesaria para el diagnóstico. La biopsia de piel, cuando se obtiene de una pústula reciente, favorece la precisión diagnóstica (9).
El diagnóstico de DPS se sospechó a partir de la experiencia de los dermatólogos que posteriormente examinaron a estos pacientes, o luego de la correlación histopatológica.
Los hallazgos histopatológicos fueron compatibles con DPS: pústulas subcórneas con acumulación de neutrófilos, en ausencia de acantólisis o espongiosis. También se observaron infiltrados dérmicos en cinco de las seis biopsias: en el caso cinco el infiltrado fue de predominio eosinofílico, en el caso seis fue de tipo linfocitario y en los demás fue de predominio neutrofílico.
En la paciente número uno se realizaron estudios de IFD, con presencia de inmunocomplejos IgA e IgG. La inmunofluorescencia usualmente es negativa en la DPS (17); sin embargo, se han descrito anticuerpos IgA en la parte superficial de la epidermis, que actúan contra la proteína desmogleína-1 (Dsg-1), hallazgo que hace parte de una variante del pénfigo por IgA, denominada tipo dermatosis pustular subcórnea (18,19).
La dapsona es el tratamiento de elección para la DPS (11,20,21). A todas las pacientes se les prescribió dapsona oral, con excepción del caso seis (Tabla 1); se observó mejoría en cuatro de ellas, con involución progresiva de lesiones y sin recurrencia después de tres meses. Solo una paciente no asistió a control y se desconoce su evolución. El mecanismo de acción de la dapsona en la DPS no está esclarecido. Se sabe que actúa inhibiendo la quimiotaxis de neutrófilos y la adherencia de polimorfonucleares al endotelio vascular (8,9). Aunque la respuesta al tratamiento suele ser rápida (entre 1 y 4 semanas), se recomienda continuar la terapia de mantenimiento (1).
La paciente de mayor edad (caso número seis) recibió tratamiento con corticoides tópicos y presentó mejoría total a los tres meses. La corticoterapia solo se recomienda en casos leves (4) y en combinación con dapsona y fototerapia (4,22), o cuando el paciente cursa, además, con otra dermatosis corticosensible (4,5). Otros tratamientos reportados en la literatura (23,24) con respuesta terapéutica variable se enumeran en la Tabla 4.
Tabla 4: Opciones terapéuticas para el tratamiento de la DPS (20,21,24)
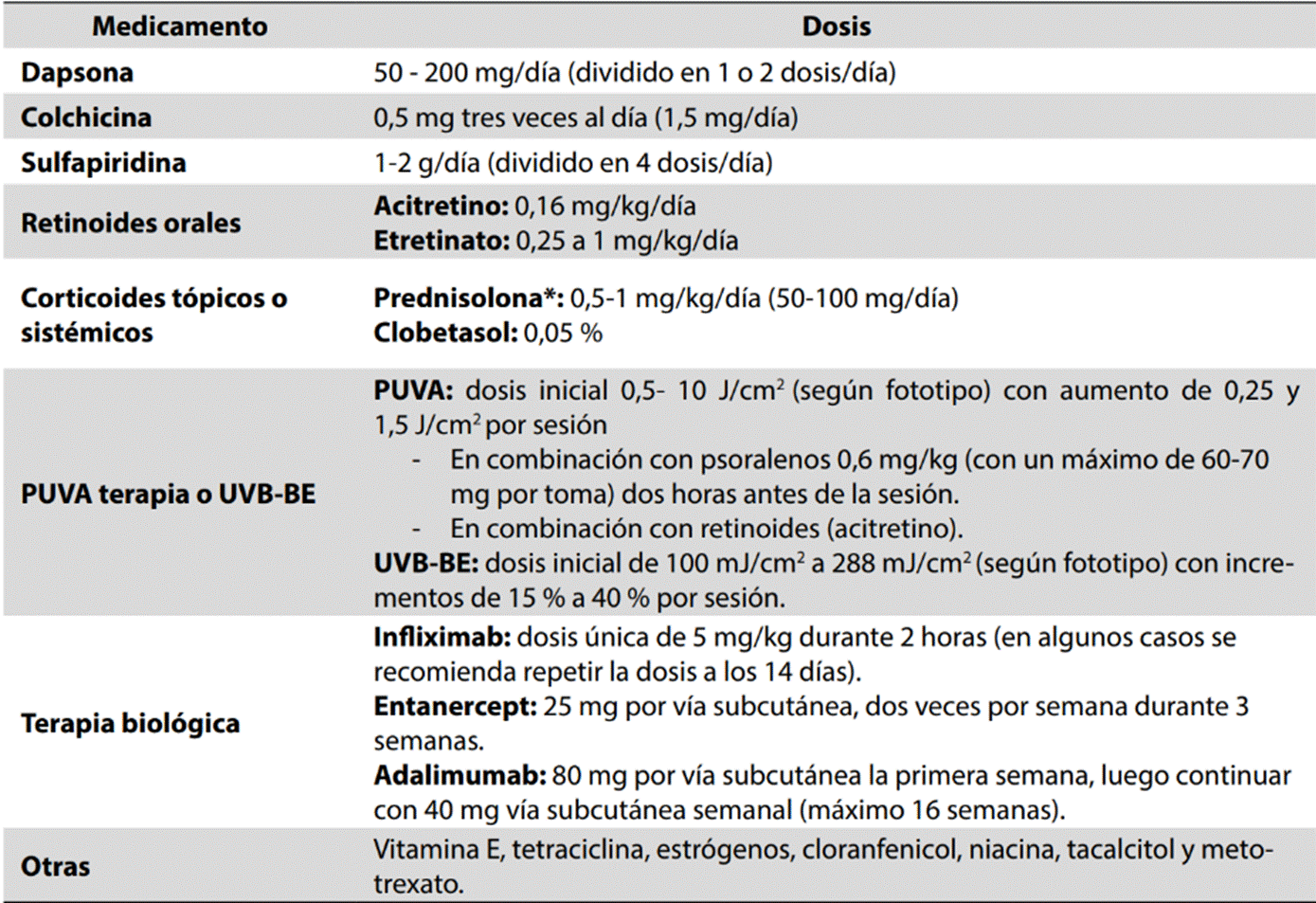
*Prednisolona: solo cuando existe otra enfermedad sistémica asociada, como pioderma gangrenoso, mieloma múltiple, etc. Fuente: creación propia
Aunque la DPS puede ocurrir en ausencia de cualquier trastorno subyacente, se ha descrito en asociación con enfermedades del tejido conectivo, enfermedad inflamatoria intestinal, disfunción tiroidea y hemopatías mieloides (24,25). Las gammapatías pueden ser el evento primario y anteceder a la DPS, ser simultáneas o aparecer tiempo después (3), como consecuencia del estímulo antigénico en la epidermis. Aunque todas las pacientes fueron estudiadas con diferentes pruebas de laboratorio, en dos de ellas se consideró realizar electroforesis de proteínas, reportadas sin pico monoclonal ni elevación policlonal de inmunoglobulinas.
La paciente con lesiones más extensas (caso número cinco) refirió astenia, mialgias, artralgias y cefalea; fue estudiada por medicina interna, reumatología y neurología, entre otras especialidades. El único hallazgo anormal fue la electromiografía de miembros superiores, que reportó una miopatía inflamatoria no clasificada. La paciente rechazó la biopsia muscular. Esta miopatía podría ser de origen autoinmune; sin embargo, tal asociación no ha sido reportada previamente en la literatura (26).
Conclusión
La experiencia adquirida durante 11 años en el diagnóstico y manejo de la DPS permitió recopilar información inédita sobre esta enfermedad, que complementa y actualiza la literatura existente. El reconocimiento de sus manifestaciones clínicas permite orientar oportunamente la sospecha diagnóstica y favorece una adecuada correlación clínico-patológica.

