











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
El carcinoma adrenal es una neoplasia endocrina poco frecuente con una incidencia reportada entre 0.7 a 2 casos por millón, pero altamente agresiva, con una supervivencia a 5 años menor al 25% para los pacientes con tumores en estadios TNM III y IV. Puede aparecer en cualquier edad, con una incidencia mayor entre los 40 a 50 años y se presenta con más frecuencia en mujeres en una razón de 5:1. Hasta el 60% pueden ser secretores, siendo el cortisol la hormona más frecuentemente secretada 1,2. La supervivencia global de estos pacientes parece estar principalmente impactada de manera desfavorable por características histológicas del tumor, tales como el índice de proliferación 3, el compromiso vascular 4,5, la extensión de la enfermedad basada en la estadificación tumoral 2,5 y la evidencia de secreción hormonal 5,6. Por el contrario, la extirpación completa del tumor 7 y el uso de Mitotane adyuvante 8 han mostrado un impacto favorable sobre los desenlaces clínicos. Sin embargo, la relación de estas variables con la supervivencia global no ha sido consistente en las diferentes cohortes publicadas.
El conocimiento de los factores que impactan el pronóstico de supervivencia ha generado cambios en el tratamiento de los pacientes, pero debido a la baja frecuencia de presentación, la experiencia de centros de referencia sigue siendo fundamental para caracterizar la población de pacientes con esta patología. En este estudio se describen las características clínicas e histológicas de los pacientes con carcinoma adrenocortical tratados en el Instituto Nacional de Cancerología (INC) y los desenlaces de supervivencia global y supervivencia libre de recaída.
Metodología
Se incluyeron pacientes mayores de 18 años, con diagnóstico histológico de carcinoma adrenal, que recibieron tratamiento quirúrgico o médico en el INC entre el 1 de enero de 2007 y 31 de diciembre de 2017. Todos los pacientes debían tener un seguimiento en el INC de mínimo de 6 meses luego de la cirugía o del inicio del tratamiento sistémico. Se excluyeron aquellos con pérdida de seguimiento mayor a 2 años continuos. Se realizó la descripción de variables clínicas e histopatológicas tales como el compromiso capsular, compromiso vascular microscópico, el índice mitótico medido en 50 campos de alto poder, el índice de proliferación medido por la expresión en inmunohistoquímica de Ki67 expresado en porcentaje. Se tuvo en cuenta la estadificación TNM de la Red Europea para el estudio de los tumores adrenales, ENSAT, por sus siglas en inglés 8.
El análisis descriptivo de las características incluyó la estimación de proporciones para las variables categóricas, y medidas de tendencia central (promedios, medianas, desviaciones estándar y rangos) para las variables numéricas. Para las estimaciones de la supervivencia global, supervivencia libre de progresión y supervivencia libre de recaída, se empleó el método de Kaplan-Meir. La supervivencia global fue definida como el tiempo trascurrido en meses entre la fecha de la cirugía de adrenalectomía para los pacientes operados o la fecha de inicio de quimioterapia para los metastásicos no operados, y la fecha de muerte o el último seguimiento. La supervivencia libre de progresión se calculó entre el inicio de la terapia sistémica y el evento de progresión (crecimiento de la suma de los diámetros de las lesiones blanco, mayor al 20%). La supervivencia libre de recaída se calculó en los pacientes sometidos a tratamiento quirúrgico con resección completa (R0), analizando el tiempo entre la cirugía y el evento de recaída estructural confirmada por imágenes de tomografía computarizada o resonancia magnética. Se llevaron a cabo análisis bivariados entre las variables de sexo (hombre vs mujer), lateralidad del tumor primario (derecha vs izquierda), secreción hormonal (sí vs no) ni estadio (I y II vs III y IV) y la mediana de la variable ki67, en relación con los desenlaces de progresión o recaída (presencia vs ausencia) y muerte (vivo vs muerto) utilizando las pruebas de Ji cuadrado y Fisher para las variables categóricas y las pruebas T o Mann Withney en el caso de variables continuas. El nivel de confianza para todos los cálculos fue del 95%. Los análisis se realizaron con SPSS Vr. 17.
Resultados
Se estudiaron las historias clínicas de 25 pacientes con diagnóstico de carcinoma adrenocortical, de las cuales 21 reunieron los criterios de inclusión, pero se excluyeron 2 porque la confirmación del diagnóstico histopatológico ocurrió por fuera del tiempo establecido en el protocolo. De los 19 pacientes incluidos, 14 fueron mujeres y la edad media al diagnóstico fue de 43.4 años (rango 20 - 65).
Las características de los pacientes se resumen en las Tablas 1 y 2.
Tabla 2 Análisis bivariado para progresión.
| Variables | Valor de p |
|---|---|
| Sexo (Mujer/Hombre) | 1.000 |
| Lateralidad (Izq/Derecha) | 0.633 |
| Funcionalidad (No/Si) | 0.377 |
| Ki 67 (<10 / >10) | 0.307 |
| Estadio (I-II / III-IV) | 0.633 |
Once pacientes presentaron secreción hormonal, siendo la producción de cortisol la predominante (n=8, 73%), 3 de ellos con secreción mixta de cortisol y andrógenos. Los pacientes con tumores secretores se distribuyeron de manera similar entre aquellos con enfermedad localizada (estadios I y II) o avanzada (estadios III y IV) al momento del diagnóstico.
Para el tratamiento, 18 pacientes fueron llevados a cirugía, y la mayoría de dichas intervenciones se realizaron en otras instituciones (13 pacientes, 72,2%). Doce pacientes fueron operados con intención curativa (estadios I a III), 3 de ellos operados por laparoscopia (tamaño tumoral menor de 6cm en 2 de ellos). De estos 12 pacientes, se logró un estado postquirúrgico R0 en 8 (66%). Tres pacientes fueron clasificados como R2 y uno como R1. Entre los 8 pacientes considerados R0, 3 presentaron recaída con un tiempo promedio al evento de 27,4 meses (14,5 a 50). Se empleó radioterapia en el lecho quirúrgico en 3 de los 12 pacientes llevados a cirugía con intención curativa (25%) y 7 pacientes tuvieron indicación de terapia adyuvante (1 por estadio III y 6 por Ki67 mayor a 10%), de los cuales 6 recibieron Mitotane. Dos de los 6 pacientes con Mitotane adyuvante presentaron recaída, con una media de 16,1 meses (rango 15,5 a 16,7).
Se encontró compromiso metastásico al diagnóstico (estadio IV) en 7 pacientes (36,8%) y 3 pacientes adicionales presentaron metástasis durante el seguimiento. Todos los pacientes con metástasis, excepto 2, recibieron quimioterapia empleando el esquema Etopósido Doxorrubicina Cisplatino (EDP) + Mitotane en 5 de ellos (50%) y EPD sin Mitotane en 3 pacientes (30%). La mejor respuesta observada fue de enfermedad estable, con un tiempo promedio de supervivencia libre de progresión de 18 meses +/-7.86 (Figura 1). Solamente una paciente en estadio IV tuvo respuesta parcial que le permitió ser llevada a cirugía posteriormente, quedando con enfermedad residual de pequeño tamaño, tratada con Mitotane por más de 12 meses. El esquema de quimioterapia empleado como segunda línea ante la progresión fue gemcitabina/capecitabina. Se encontró una diferencia significativa entre la mediana del valor del índice de proliferación Ki67 entre los paciente que no presentaron progresión o recaída (15,0% rango 4 - 20) y los que sí (20,0%, rango 1 a 90) p=0.01. No hubo diferencias para las variables de sexo (hombre vs mujer), lateralidad del tumor primario (derecha vs izquierda), secreción hormonal (si vs no) ni estadio (I y II vs III y IV) .
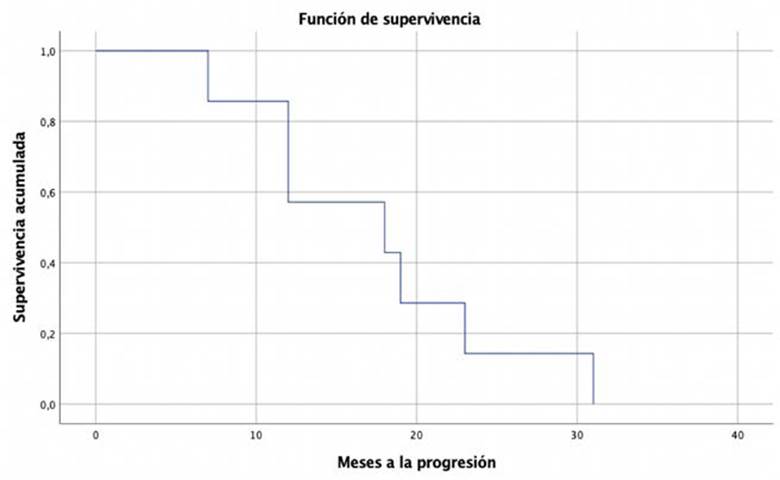
Al final del seguimiento, 8 pacientes habían fallecido: 4 de 7 (57,1%) pacientes en estadio IV, 1 de 4 (25%) en estadio III y 3 de 7 (42,8%) en estadio II. El tiempo medio de supervivencia global fue de 30 meses +/-19.80 (Figura 2). En el análisis bivariado se encontró una diferencia significativa entre la mediana del valor de Ki67 entre los pacientes que permanecieron vivos (16,5, rango 1 - 20) y los que fallecieron (19,04, rango 2 a 90) p=0.186. No hubo diferencias para las variables de sexo (hombre vs mujer), lateralidad del tumor primario (derecha vs izquierda), secreción hormonal (si vs no) ni estadio (I y II vs III y IV).
Discusión
El carcinoma adrenal es una neoplasia rara pero altamente agresiva. Se presenta en todas las edades pero es más frecuente entre los 40 a 50 años. Es ligeramente más prevalente en las mujeres sin llegar a duplicar la prevalencia en hombres 2,3,9,10 lo cual contrasta con esta serie en la cual se encontró una relación mujer: hombre de 2.8:1, similar a lo encontrado en una gran serie brasilera 5. Respecto a la lateralidad, se encontró una ligera mayor frecuencia de casos en la suprarrenal derecha en contraste con otros estudios con mayor número de pacientes 2,5,10 pero similar a un estudio publicado recientemente en India 11 en el cual identificaron lateralidad derecha en 21 pacientes vs lateralidad izquierda en 16 pacientes de la cohorte.
La evaluación bioquímica previa a la cirugía en pacientes con masas adrenales es importante y los expertos recomiendan hacerlo de manera rutinaria (13,14. En esta serie se encontraron 11 pacientes con tumores secretores, siendo el cortisol la hormona más frecuentemente secretada (8 pacientes), en concordancia con lo descrito en la literatura 5,12,15. Frente al papel de la secreción hormonal como variable para determinar el riesgo de recurrencia o de metástasis, en la literatura hay evidencia contradictoria; por ejemplo, en un estudio de la región 5 la secreción de hormonas se asoció a un peor pronóstico, pero otros estudios no han demostró dicha asociación 10,12. En un estudio reciente de Mayo Clinic se encontró una supervivencia general mejor para los carcinoma no funcionales (mediana de supervivencia 66 vs 22 meses, p.01), HR 1.5 (95%IC 1.04-2.14) comparada con no secretores, sin diferencias entre la morbilidad a 30 días ni el estado postquirúrgico de resección completa (R0) 16.
El tamaño tumoral no tiene una evidencia sólida a favor o en contra de su asociación como factor pronóstico. En uno de los estudios 3, en el análisis multivariado se atribuyó una tasa de riesgo (HR) de 1.6 (IC 95% 1.03 a 2.4) para supervivencia libre de recaída a los tumores con diametros entre 15 y 20 cm, pero sin una asociación significativa con la supervivencia global. En la misma vía, el estudio de Freire 5 encontró una asociación con mal pronóstico con un tamaño tumoral mayor o igual a 7.5 cm. Sin embargo, otros estudios no han encontrado asociación con dichos desenlaces 11,12. El tamaño tumoral ayuda a definir el tipo de abordaje quirurgico, aunque ante la sospecha de malignidad se aconseja más el abordaje abierto frente al laparoscópico 17. En el presente estudio, 12 pacientes fueron intervenidos con intención curativa, logrando un estado postquirúrgico R0 en el 66% de los casos, que es una proporción más baja a la reportada en otros estudios retrospectivos 16,19, con tasas de resección R0 del 72 al 85%.
Las variables histológicas relacionadas con la proliferación celular, tales como índice mitótico y el KI 67%, tenidas en cuenta en estudios más recientes, parecen tener el mayor impacto en el pronóstico tanto para sobrevida global como para sobrevida libre de progresión 3,5. El punto de cohorte para definir un peor pronóstico no está claramente establecido y solo un estudio 15 reporta un peor pronóstico en los tumores con un conteo mitótico mayor a 20 mitosis/50 CAP. Para el índice de proliferación determinado por KI67%, las guías de la Sociedad Europea de Oncología 17 definieron un punto de corte mayor al 10% como criterio de indicación de adyuvancia con Mitotane, y ésta recomendación fue reforzada por los hallazgos de otros investigadores 3,20,21. En este estudio, la mediana de Ki67 fue diferente en los grupos de fallecidos vs vivos y aquellos con recaída/progresión frente a no recaída/progresión, a pesar de la escasa muestra del estudio.
La terapia adyuvante con Mitotane está indicada según las recomendaciones actuales 1,18,20 en pacientes con probable enfermedad residual microscópica (Rx o R1) o con estadificación III de ENSAT o con KI67% mayor al 10%. En esta serie, se encontraron 7 pacientes con indicación de terapia adyuvante de los cuales 6 recibieron Mitotane. Cuatro de ellos continuaron libres de recaída y solamente 2 recayeron con una media de tiempo a la recaída de 16,1 meses (rango 15,5 a 16,7). Por el pequeño tamaño de la muestra no se encontraron diferencias en la supervivencia libre de recaída ni en la supervivencia global.
La supervivencia global a 5 años está asociada con el estadio tumoral de la Red Europea para el estudio de los Tumores Adrenales - ENSAT 8, siendo del 66% al 82% para el estadio I, del 58% al 64% para el estadio II, del 24% al 50% para el estadio III y tan solo del 0% a 17% para el estadio IV 9. En este estudio no fue posible estimar la tasa de supervivencia global a 5 años estratificada por estadio ENSAT dadas las limitaciones de la muestra.
Conclusión
En esta serie de casos se caracterizaron los pacientes con carcinoma adrenal tratados con cirugía o manejo médico en una institución oncológica en un periodo de 11 años. Se destaca la menor frecuencia de resecciones R0 en los pacientes llevados a cirugía con intención curativa, comparado con los datos de otras series, posiblemente debido a que la mayor parte de los pacientes fueron operados en centros no oncológicos y luego remitidos para continuar tratamiento y seguimiento. También se resalta la adherencia a las guías ESMO en cuanto a los esquemas de adyuvancia y quimioterapia, y los desenlaces observados en supervivencia global y supervivencia libre de progresión, que son comparables a los publicados por instituciones con alta experiencia en patología adrenal. Se concluye con esto, que debe procurarse el tratamiento de pacientes con carcinoma adrenocortical en instituciones que puedan ofrecer experticia y manejo multidisciplinario

