










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Química
Print version ISSN 0120-2804
Rev.Colomb.Quim. vol.43 no.3 Bogotá Sept./Dec. 2014
Cristalización y predicción de la estructura tridimensional de la proteína homóloga del receptor activado para la quinasa c (lack) de leishmania
Crystallization and prediction of three-dimensional structure of the homologue protein of the receptor for activated c-kinase (lack) from leishmania
Cristalização e predição da estrutura tridimensional da proteína homologa do receptor ativado para a quinasa c (lack) de leishmania
Edwin Patiño-González1*, Isabel Andrea Patiño-Márquez2, Juan Fernando Alzate3
1 Profesor Asistente, Grupo de Bioquímica Estructural de Macromoléculas, Instituto de Química, Universidad de Antioquia, Medellín, Colombia. e-mail: edwin.patino@udea.edu.co.
2 Estudiante PhD, Grupo de Bioquímica Estructural de Macromoléculas, Instituto de Biología, Universidad de Antioquia, Medellín, Colombia. e-mail: isabel.patino@udea.edu.co.
3 Profesor Asociado, Grupo de Parasitología, Departamento de Microbiología y Parasitología de la Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia. e-mail: jfernando.alzate@udea.edu.co.
* Autor de correspondencia: Edwin Patiño González, dirección: Calle 67, No 53-108, email: edwin.patino@udea.edu.co, teléfono: 219 56 52 ó 219 56 62; celular 301 767 8473.
Article citation:
Gónzales-Patiño, E.; Patiño-Márquez, A. I.; Alzate, J. F. Cristalización y predicción de la estructura tridimensional de la proteína homóloga del receptor activado para la quinasa C (lack) de Leishmania. Rev Colomb Quim. 2014. 43(3): 17-23.
Recibido: 21 Agosto 2014. Aceptado: 25 de septiembre 2014
Resumen
Los parásitos del género Leishmania son los causantes de la enfermedad conocida como Leishmaniasis. Esta enfermedad es endémica en 98 países. Veinte especies de Leishmania sp han sido descritas como patógenos en humanos y varias de ellas presentan manifestaciones clínicas diferentes. No se dispone de vacuna, a pesar del considerable esfuerzo de muchos grupos de investigación. Las alternativas para descubrir nuevos medicamentos están basadas en el diseño de compuestos que interaccionen con blancos específicos, principalmente, proteínas encargadas de procesos metabólicos o celulares del patógeno, e.g. la parasitación de las células del huésped vertebrado. La eficiente parasitación del huésped vertebrado por Leishmania depende de la expresión de diferentes proteínas, incluyendo la proteína Lack. Parásitos deficientes de Lack no sobreviven internalizados en las células de los vertebrados. Este artículo presenta las condiciones de renaturalización, purificación y cristalización de la proteína Lack del patógeno humano Leishmania (Viannia) panamensis. Además, los resultados de modelación estructural de esta proteína muestran una conformación proteica similar a un ventilador organizado en 7 aspas, cada una compuesta de 4 hojas β. La estructura de la proteína Lack resultó similar a la proteína asociada a ribosoma RACK1 de Trypanosoma brucei y Saccharomyces cerevisiae, y a la de otros eucariotas. Las características estructurales de la proteína Lack podrían ser usadas para la exploración de nuevos.
Palabras claves: Cristalización, estructura, Lack, Leishmania, renaturalización.
Abstract
Leishamnia parasites are the causative agents of the leishmaniasis disease. Due to its broad distribution, parasites are endemic in approximately 98 countries. Twenty species of Leishmaniasp has been described as human pathogens and several of them present different clinical manifestations. This feature poses a significant challenge to the general goals of parasite control and erradication. There is no a protective vaccine for humans, despite substantial efforts by many research teams. Alternatives to discover new drugs are based on the design of new compounds that bind selected targets. Mainly, the targets are proteins involved in key metabolic or cellular processes of the pathogen, e.g. parasitization of vertebrate host cells. The efficient parasitization of the vertebrate host by Leishmania parasites depends on the expression of different molecules including Lack protein. The knockout parasites fail to survive inside the vertebrate host cells. In this article we highlight the conditions to perform the refold, purification, and crystallizing of the Lack protein of the human pathogen Leishmania (Viannia) panamensis. Moreover, we present structure modelling analysis which shows a protein conformation like a fan organized in 7 blades, each one composed of 4 b sheets. Furthermore, the structure of Lack protein was found to be similar to RACK1-ribosome associated protein from Trypanosoma brucei and Saccharomyces cerevisiae and other eukaryotes. The structural characteristics of Lack protein could be used for exploration of new drugs.
Key words: Crystallization, Lack, Leishmania, refolding, structure.
Resumo
Os parasitas do gênero Leishmania são os agentes causadores da doença conhecida como Leishmaniasis. Esta doença é endêmica em 98 países. Vinte espécies de Leishmania sp têm sido descritas como patógenos em humanos e várias delas apresentam manifestações clínicas diferentes. Não se dispõe de vacina, apesar do considerável esforço de muitos grupos de pesquisa. As alternativas para descobrir novos medicamentos estão baseadas no desenho de compostos que interajam com alvos específicos, principalmente, proteínas encarregadas de processos metabólicos ou celulares do patógeno, e.g. a parasitação das células do hóspede vertebrado. A eficiente infestação do hóspede vertebrado por Leishmania depende da expressão de diferentes proteínas, incluindo a proteína Lack. Parasitas deficientes de Lack não sobrevivem internalizados nas células dos vertebrados. Este artigo apresenta as condições de regeneração, purificação e cristalização da proteína Lack do patogénico humano Leishmania (Viannia) panamensis. Além disso, resultados de modelação estrutural desta proteína mostram uma conformação protéica similar a um ventilador, organizado em 7 pás, cada uma composta de 4 folhas β. A estrutura da proteína Lack resultou similar a proteína associada ao ribossomo RACK1 de Trypanosoma brucei y Saccharomyces cerevisiae, e à de outros eucariotas. As características estruturais da proteína Lack poderiam ser usadas para a pesquisa de novos fármacos.
Palavras chave: cristalização, estrutura, Lack, Leishmania, renaturalização.
Introducción
La leishmaniasis es una enfermedad parasitaria endémica en aproximadamente 98 países (1). El parásito que causa la enfermedad es un protozoario flagelado que puede infectar una variedad de mamíferos y que es transmitido al humano a través de la picadura de un insecto vector (2). Los humanos infectados con el parásito de Leishmania padecen de diferentes manifestaciones clínicas que van desde ulceras localizadas hasta infecciones viscerales (3). Kelly y colaboradores (4) investigaron el ciclo de vida de Leishmania y la forma en la que este mantiene su viabilidad mientras se encuentra dentro de células del sistema inmune humano, como los macrófagos. Sus resultados mostraron que uno de los factores que modulaba la viabilidad intracelular del parásito era la expresión del gen homólogo del receptor de la quinasa C activada (Lack) de Leishmania. Los resultados con parásitos que carecían de la expresión de Lack (Lack null Leishmania) demostraron que estos fueron incapaces de establecer una infección intracelular, o mantenerse intracelularmente después de la inoculación en ratones inmunodeficientes. Además, parásitos con expresión de solo una copia del gen Lack mostraron una replicación atenuada en macrófagos. En otro estudio, se encontró que el gen Lack de Leishmania codifica para una proteína que se une con el plasminógeno del huésped para facilitar la invasión del parásito a los macrófagos humanos (5). Los resultados encontrados por Kelly, Gómez-Arreaza y sus respectivos colaboradores (4, 5) sugieren que el diseño de sustancias capaces de disminuir o inhibir la actividad de Lack, puede ser eficaz en el tratamiento de la Leishmaniasis. Descifrar los aspectos estructurales de la proteína Lack permitiría comprender la relación estructura actividad de la proteína y su función molecular en el parásito. La predicción de estructuras por medio de programas computacionales se ha convertido en una excelente aproximación para el campo de la biología estructural (6, 7) cuando no se dispone de estructuras obtenidas por cristalografía o por resonancia magnética nuclear (RMN). La predicción permite entender los procesos bioquímicos de las moléculas biológicas llevando los análisis computacionales a nivel del tubo de ensayo. En la actualidad, diferentes programas de computación pueden predecir la estructura tridimensional (3-D) de cualquier proteína (6, 8, 9). Este artículo muestra por primera vez las condiciones de cristalización de la proteína Lack como paso previo a la determinación experimental de su estructura por cristalografía. Además, se reporta la predicción de un modelo estructural de Lack que permite definir los segmentos que adoptan una estructura local regular como la hoja plegada-b de sus 312 aminoácidos.
Materiales y métodos
Clonación de la secuencia de Lack
La secuencia que codifica para la proteína Lack fue amplificada del DNA genómico de L. (V.) panamensis cepa UA946 (Universidad de Antioquia). La reacción en cadena de la polimerasa (PCR) se realizó en 25 μl de volumen final que contenían: dNTPs 0,2 mM, 1X tampón para Taq-polimerasa (Tampón: 10X: Tris-HCl 100 mM pH 8,8, KCl 500 mM y 0,8 Nonidet P40), 0,5 U de la enzima Taq-polimerasa, 0,25 μM del cebador adelantado (sitio de restricción NdeI subrayado, CCCCATATGATGAACTACGAGGGTCACCTGAAG), 0,25 μM del cebador atrasado (sitio de restricción HindIII subrayado, CCAAGCTTCTCGGCGTCCGAGATGGA), MgCl 21,2 mM y 5 ng de ADN. La amplificación contó con un ciclo de desnaturalización inicial del ADN de 1:20 min, 40 ciclos de amplificación (desnaturalización 30 s, apareamiento de cebadores 30 s y extensión del cebadores 1:30 min), y 1 ciclo final de terminación de 20 min. Los productos de la PCR fueron purificados, digeridos y clonados en el vector de expresión pET28a (Invitrogen) en los sitios de restricción NdeI y HindIII (pET28a-Lack). Este constructo que codifica para los residuos Met1 hasta Glu312 de la proteína Lack fue obtenido en el Centro Nacional de Secuenciación Genómica por el Dr. Juan Fdo Alzate (CNSG, Universidad de Antioquia). El producto final contiene dos secuencias His-Tag a cada extremo de la proteína. Además, un sitio para corte con trombina y un T7-Tag, ambos localizados en el amino terminal. Estas secuencias pertenecientes al vector pET-28a incrementan el número de residuos de 312 a 346, lo que representa un peso molecular estimado de 38,1 kDa. La secuencia de Lack fue verificada por secuenciamiento capilar según el método de Sanger por la compañía Macrogen (Seúl, Korea).
Preparación de Lack de L. (V.) panamensis
Células de E. coli BL21 (DE3) fueron transformadas con el vector pET28a-Lack por choque térmico. El cultivo preparativo en medio Luria Bertani (LB) suplementado con kanamicina (50 mg/ml) fue preparado a 37 ºC con agitación a 150 rpm hasta que alcanzó una densidad óptica (OD600) de 0,5 a 600 nm. La expresión de la proteína por parte de las células transformadas con el vector de expresión pET28a-Lack fue inducida con IPTG a una concentración final de 1 mM. Luego de la inducción, las células fueron dejadas por 4 h más a las mismas condiciones de temperatura y agitación. Finalmente, las células fueron concentradas por centrifugación a 5000 F.C.R.(fuerza centrífuga relativa)por 15 min a 4 ºC. Las células fueron resuspendidas en 10 volúmenes de tampón de lisis TBSE (Tris-HCl 10 mM pH 8,0, EDTA 1,0 mM, NaCl 150 mM, y β-ME (β-mercaptoetanol) al 1 % v/v. Se agregaron a la suspensión 50 µL de un coctel de inhibidores de proteasas (Calbiochem) para prevenir la degradación proteolítica de Lack. Las células fueron lisadas por ultrasonido con una frecuencia de 20 kHz, una amplitud de 40 % y con una punta de titanio de 6 mm de diámetro (Sonics, VCX 750), durante 5 min en baño de hielo. La mezcla fue centrifugada a 8500 F.C.R. por 30 min a 4 ºC para separar la fracción soluble (sobrenadante) de la insoluble (Cuerpos de Inclusión, CI). Los CI fueron lavados dos veces con tampón TBSE. La proteína acumulada en los CI fue solubilizada con 5 volumenes de tampón de solubilización (Tris 10 mM pH 8,0, EDTA 1 mM y GuHCl 8 M (Cloruro de Guanidinio)). Luego, fue incubada por 12 h a temperatura ambiente. Posteriormente, la suspensión fue centrifugada a 9500 F.C.R. por 30 min a 4 °C. La concentración de proteína se determinó por OD280 utilizando el coeficiente de extinción (2,12 mg/ml). La proteína se concentró a 20 mg/ml en una celda con agitación (Millipore) utilizando membranas YM3 con un tamaño de poro de 3000 Da (MWCO 3000).
Purificación y renaturalización de la proteína Lack
La proteína Lack extraída de los CI y concentrada a 20 mg/ml en presencia de GuHCl 8 M se cargó en una columna de Ni (Profinity, BioRad) preequilibrada con GuHCl 6 M e imidazol 10 mM. La proteína unida a la columna se eluyó con el tampón de GuHCl 6 M y de imidazol 250 mM. Se tomaron fracciones de 2 ml, las cuales fueron analizadas por SDS-PAGE teñido con azul de coomassie. Las fracciones que se visualizaron con una mayor cantidad de proteína en el SDS-PAGE se mezclaron, se concentraron nuevamente hasta alcanzar una concentración de 20 mg/ml, y fueron almacenadas a -20 ºC hasta ser utilizadas para la renaturalización (conformación funcional). Para la renaturalización, la proteína Lack se diluyó a 100 mg/ml en tampón a pH 8,0 (Tris-HCl 50 mM, NaCl 100 mM, glicerol 100 mM, urea 2,0 M, L-arginina 1 M, triton X-100 1 %, b-ciclodextrina 4,5 mM, GSH 0,1 mM (glutatión reducido) y GSSH 0,01 mM (glutatión oxidado). La solución fue colocada en una membrana SpectraPor 3 de 3500 Da por 12 h a 4 ºC. La proteína empacada se dializó en 10 veces el volumen de renaturalización del tampón (Tris-HCl 50 mM pH 8,0, NaCl 100 mM, glicerol 100 mM, urea 1,0 M, L-arginina 1 M, triton X-100 1 %, b-ciclodextrina 4,5 mM, GSH 0,1 mM y GSSH 0,01 mM). Luego, la concentración de urea se redujo intercambiando cada 24 h el 50 % del tampón de diálisis con una solución de Tris 50 mM, pH 8,0. Después de 4 días de diálisis, la solución de proteína se concentró en un Amicon con celda de agitación. La proteína concentrada fue cargada en una columna de Ni pre-equilibrada con Tris-HCl 50 mM, NaCl 100 mM, b-ciclodextrina 4,5 mM y L-arginina 1 M a pH 8,0. La proteína Lack unida a la columna se purificó agregando al tampón imidazol 250 mM. Se colectaron fracciones de 1 ml, las fracciones se resuspendieron en tampón sin agente reductor (β-ME) y se analizaron por SDS-PAGE. Las fracciones que mostraron solo una banda correspondiente a la proteína Lack fueron mezcladas y concentradas por ultrafiltración hasta 1,5 mg/ml.
Análisis de pureza de la proteína Lack
El análisis de pureza de la proteína Lack se realizó en un sistema de electroforesis capilar (Bioanalyzer 2100, Agilent; Santa Clara, USA). La proteína Lack se diluyó a una concentración de 0,125 mg/ml (solución madre). Luego, 5 mL de la solución madre se mezclaron con el estuche de Agilent siguiendo las instrucciones de la casa matriz (Agilent High Sensitivity Protein 250 Kit, Cat 5067-1575). Después, la mezcla fue separada en el Bioanalyzer 2100. Los resultados fueron analizados con el software Expert package (2100 Expert package, versión B.02.07.SI532).
Cristalización de la proteína Lack
La proteína Lack fue cristalizada por el método de difusión de vapor por gota colgante empleando platos de 24 pozos (Linbro plates 24 wells), de acuerdo a un reporte previo (10). Además, se utilizó el estuche de cristalización HR2-110 que comprende 50 soluciones de cristalización numeradas del 1al 50 (Hampton Research ScreenHR2-110). Cada reacción de cristalización se preparó mezclando 2 μl de proteína (1,5 mg/ml) y 1 μl de cada solución tampón de cristalización del estuche de cristalización. La gota de cristalización fue colocada sobre 500 μl de solución madre, la mezcla resultante se dejó a temperatura ambiente por 72 h. Después de este tiempo se utilizó un microscopio óptico (Carl Zeiss Jena; NY, USA) para registrar el crecimiento de los cristales. Además, se empleó un microscopio Nikon con lente de luz polarizada.
Predicción de la estructura tridimensional (3D) de Lack
La predicción de la estructura 3D de la proteína Lack se realizó utilizando el programa I-TASSER (University of Michigan, USA). El modelo generado incluye los datos en formato PDB (Protein Data Bank). La secuencia de Lack fue extraída de nuestros resultados de secuenciación del cDNA clonado y este resultado se comparó con el archivo reportado en la base de datos GenBank (AF125254.1). La estructura generada por el programa I-TASSER involucró cuatro características principales: identificación del molde, ensamblaje de fragmentos estructurales, refinamiento atómico a nivel estructural e interpretación funcional basada en la estructura (6, 7, 9). El modelo obtenido de la estructura 3D de Lack fue optimizado con el programa ModRefiner (University of Michigan, USA) para obtener una mejor estructura tanto local como global y para minimizar las superposiciones atómicas. Por otro lado, se realizó en paralelo la predicción de estructura de Lack por el programa SAM-T08 (https://compbio.soe.ucsc.edu/SAM_T08) y se generó el archivo PDB correspondiente. Para cuantificar las diferencias entre las estructuras generadas por ModRefiner y SAM-T08 se calculó la desviación de la media cuadrática (RMSD) de los dos archivos PDB por medio del programa PyMOL (Schrödinger, San Diego, California, USA). La validación geométrica de la estructura de Lack se hizo con el programa RAMPAGE, determinando los valores de los ángulos phi (φ) y psi (ψ) (University of Cambridge, UK).
Resultados
Expresión y purificación de Lack en condiciones desnaturalizantes
La secuencia codificante del gen de la proteína Lack de L. (V.) panamensis fue amplificada por PCR y posteriormente clonada en el vector de expresión procariota pET28a. Este constructo fue confirmado mediante secuenciamiento capilar de ambas cadenas por el método de Sanger (resultados no mostrados). La expresión de Lack fue inducida en la fase exponencial del cultivo (DO600 - 0,5) y la proteína se acumuló abundantemente en la fracción insoluble, que contenía los CI de la bacteria (Figura 1A, carril 3). La proteína Lack fue visualizada mediante electroforesis SDS-PAGE como una banda de aproximadamente 38 kDa. La extracción de la proteína de los CI se realizó por solubilización con cloruro de guanidinio (GuHCl). Después de eliminar la mayoría de restos celulares por medio de centrifugación, el sobrenadante fue aclarado por filtración. Gran parte de las proteínas contaminantes fueron removidas por cromatografía de afinidad (Ni-Profinity) usando condiciones desnaturalizantes (GuHCl 6 M) (Figura 1B, carril 4).

Renaturalización y purificación de Lack en su forma nativa
La proteína desnaturalizada que eluyó de la columna de Ni con GuHCl fue plegada a su forma nativa cambiando el agente caotrópico fuerte (GuHCl) por uno más débil (urea), el cual fue diluido secuencialmente mediante un proceso de diálisis que disminuía en un 50 % la concentración de urea cada 24 h. La suplementación del tampón de diálisis con diferentes aditivos evitó que la proteína Lack precipitara. Por ejemplo, con el detergente (Triton X-100), con sales como el cloruro de potasio (KCl) y manteniendo la urea a una concentración mínima de 0,5 M se pudo tener la proteína en solución. Obtener la proteína Lack en un tampón con concentraciones más bajas de urea requirió del uso de b-ciclodextrina y L-arginina; con estos dos nuevos aditivos y con una concentración final de urea de 0,125 M la proteína se mantuvo soluble durante todo el proceso de diálisis. La proteína Lack fue finalmente purificada en una columna de Ni. El análisis de la proteína plegada por SDS-PAGE en condiciones no reductoras (tampón sin β-ME) mostró un patrón de migración más rápido que la proteína desnaturalizada (Figura 1B, carril 5).
Análisis de pureza de la proteína Lack
La cuantificación de la pureza de la proteína Lack se realizó por medio de electroforesis capilar (Bioanalyzer 2100, Agilent). Los electroferogramas obtenidos (Figura 1C) muestran un primer pico correspondiente al marcador interno del kit de Agilent a 5 kDa (marker). Dos picos más son observados, uno mayoritario de 34,1 kDa (97%) y que corresponde a la proteína Lack monomérica; y otro de una masa molecular de 67,3 kDa que representa del 2–3 % de la proteína total, y que corresponde a la proteína Lack agregada.
Cristalización de la proteína Lack
La proteína Lack con una pureza aproximada del 97 %, como se evidencia por electroforesis capilar, se concentró hasta una concentración de 1,5 mg/mlusando una celda con agitación (Millipore) y membranas de 3000 MWCT. Luego, se cristalizó por el método de la gota colgante a temperatura ambiente. El crecimiento de los cristales se encontró a las 72 h (Figura 2). Los cristales crecieron individuales en la gota, y presentaron tamaños geométricos de 200 x 200 x 50 μm. Las condiciones de cristalización del tampón # 35 del kit de cristalización presentaron los mejores resultados (Hepes sodio 0,1 M y fosfato de amonio monobásico 0,8 M, pH 7,5) (Figura 2). La evaluación de birefringencia con luz polarizada demostró que los cristales desviaban o desdoblaban la luz polarizada, lo que demuestra que la proteína cristalizó de una forma ordenada.

Predicción de la estructura tridimensional de Lack
Los avances en programas computacionales, aunado con el aumento de las estructuras reportadas en la bases de datos del PDB (www.rcsb.org) hacen posible predecir modelos cada vez más confiables de la estructura tridimensional de una proteína partiendo solo de su estructura primaria. Considerando que la estructura de la proteína Lack de L. (V.) panamensis es desconocida se generó un modelo utilizando el software I-TASSER. El modelo 3D obtenido resultó ser satisfactorio (C-score 1,55), y la topología de la proteína resultó correcta (TM-score 0,93 ± 0,06), por lo que la estructura de la proteína Lack puede considerarse, con un alto nivel de confianza, como un polipéptido conformado por 17 hojas-b organizadas de forma circular. El modelo 3D de la estructura presenta un diámetro promedio de 43 Å y una altura de 35 Å. La forma circular observada en Lack, hace que se forme una abertura de 11 Å en el centro de la molécula, lo que supone el transporte de moléculas o la posibilidad de interacción con posibles ligandos (Figura 3). La proteína presenta 68 residuos estructurados en una región flexible de lazo (loop), que se extiende desde el residuo 1-68 en la parte amino terminal. La región entre los residuos 69-190 está compuesta por 12 hojas-b antiparalelas conectadas por lazos. Los residuos 191-256 se organizan en la segunda extención flexible (lazo) de la proteína (Figura 3A, parte superior). Dos nuevas hojas-b conectadas por lazos se encuentran entre los residuos 257-270. Los últimos 36 residuos de la parte carboxi terminal forman 3 hojas-b antiparalelas. La estructura de Lack presenta una estructura asimétrica, en la cual se distingue una parte con 12 hojas-b antiparalelas (mitad izquierda), mientras la otra parte consta de regiones desorganizadas con solo 5 hojas-b antiparalelas (mitad derecha) (Figura 3, parte superior). La estructura fue optimizada utilizando el programa ModRefiner que reduce las difierencias estructurales entre nuestro modelo con la(s) estructura(s) de alta resolución depositada(s) en el portal de estructuras biológicas macromoleculares (PDB). El refinamiento de la estructura inicial de Lack aumentó el número de hojas-b antiparalelas de 17 a 28 (Figura 3B, parte inferior). La primera región flexible que se extendía desde el NH2 terminal hasta Val 68 se plegó en 5 nuevas hojas-b. De la misma forma, la segunda región flexible que comprendía desde la Lys 191 hasta la Lys 256 se plegó en 6 nuevas hojas-b. La estructura final de Lack se asemeja a una turbina con 7 aspas y cada aspa está formada por 4 hojas-b antiparalelas (Figura 3B, parte inferior).

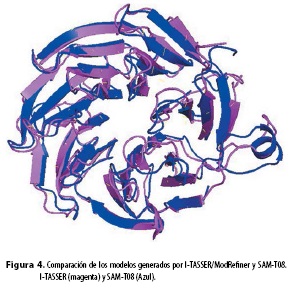
La determinación de los valores de ϕ y ψ con el programa RAMPAGE dio como resultado la distribución de los residuos de la proteína Lack en el gráfico de Ramachandran. La estructura presenta 295 residuos (95,2%) en la región más favorecida, 13 residuos (4,2%) en regiones permitidas y 2 residuos (0,6%) en regiones atípicas (resultados no mostrados). Por otro lado, la determinación de la estructura de Lack con el programa SAM-T08 mostró la misma organización estructural global que la obtenida por I-TASSER (resultados no mostrados). Las dos estructuras (archivos PDB) se solaparon casi perfectamente con el programa PyMol (Figura 4), con una RMSD de 1,12 Å. Algunos residuos muestran diferencias menores en el solapamiento que probablemente tienen efectos menores en la estructura (Figura 4).
Discusión de resultados
Muchas de las proteínas heteorólogas expresadas en E. coli son acumuladas en los cuerpos de inclusión, por lo que su recuperación precisa de un paso de renaturalización. Este proceso requiere del uso de aditivos como detergentes, sales, alcoholes, etc., en las soluciones tampón de plegamiento. No obstante, en nuestro caso, el detergente no iónico Triton X-100 no produjo un aumento de la solubilidad de Lack. Además, aumentar las interacciones electrostáticas con bajas concentraciones de sales (e.g., KCl) no fue suficiente para mantener la proteína soluble, por lo que más del 95 % de la misma precipitó en la membrana de diálisis. Posiblemente la causa de la agregación de los monómeros durante la renaturalización fue por la oxidación de las cisteínas para formar cistina (11). La agregación y precipitación de Lack fue disminuida eficientemente adicionando al tampón de renaturalización L-arginina y b-ciclo dextrina. Aunque el papel de la L-arginina no es totalmente conocido, se presume que este amino ácido rodea las regiones hidrofóbicas parcialmente plegadas, y así disminuye la probabilidad agregación (12-14). Por otro lado, se ha visto que la b-ciclo dextrina forma complejos con el detergente removiéndolo del complejo proteína/detergente, y de esta forma facilita la renaturalización de los polipéptidos (15). Nuestros resultados muestran claramente que la adición de L-arginina 1 M de y de b-ciclo dextrina 4,5 mM favorecen el proceso de renaturalización de Lack manteniendo la proteína soluble. El análisis en SDS-PAGE mostró que la proteína plegada tuvo un patrón electroforético más rápido que la proteína desnaturalizada, este cambio en el patrón de migración puede deberse a la presencia de β-ME en la proteína desnaturalizada. La proteína Lack posee 8 cisteínas en su secuencia, estos 8 grupos sulfhidrilo están distantes y no hay reportes de que formen enlaces disulfuro entre ellos, sin embargo, los grupos tiol de las proteínas recombinantes pueden formar aductos con agentes reductores como el β-ME y de esa manera, pueden cambiar su peso molecular (16).
El parámetro crucial para cristalizar una proteína es su pureza, pues este proceso depende, primordialmente, de la homogeneidad de la molécula (17, 18). Los cristales fueron obtenidos de una proteína altamente homogénea (≈97%) como lo demostraron los análisis de electroforesis capilar (Figura 1C). Aunque el tamaño de la proteína Lack obtenida varió un 10,5 % con respecto al valor esperado (de 38,1 kDa a 34,1 kDa), esta variación está dentro de las especificaciones técnicas del análisis, en donde se reporta que el tamaño del analito puede variar hasta un 20 % en el equipo Bioanalyzer 2100 de Agilent (19). Además, es probable que los agregados que se detectaron por electroforesis capilar y que no fueron visualizados por SDS-PAGE cuando se corrió la proteína en condiciones no reductoras se deban a las diferencias de resolución de ambas técnicas.
Los cristales formados por macromoléculas como las proteínas exhiben propiedades ópticas diferentes a los reactivos de cristalización utilizados, y es precisamente por esta razón que se requiere la evaluación con luz polarizada para seleccionar y diferenciar los cristales formados por proteínas de aquellos formados por los reactivos de cristalización (20). Las proteínas desnaturalizadas no forman cristales regulares debido a que la gran diversidad de formas y plegamientos presentes en solución impiden el proceso de cristalización. Nosotros pudimos obtener cristales de la proteína Lack los cuales mostraron fluorescencia cuando fueron observados con luz polarizada (Figura 2). La disponibilidad de estos cristales provee una base para el discernimiento estructural de la proteína Lack por métodos de difracción de rayos-X, y una mejor comprensión de los mecanismos de parasitación del huésped (4) y/o unión a plasminógeno (5).
Se realizó una primera aproximación a la estructura de la proteína Lack de Leishmania por medio de la modelación con el software I-TASSER (6). El modelo refinado de I-TASSER se comparó con el modelo generado por el software SAM-T08 (datos no mostrados) (8) y el resultado de RMSD fue de 1,12 Å, lo que supone que los dos modelos son similares (Figura 4). Además, en el modelo refinado los valores de los ángulos φ y ψ para el 95,2 % de los residuos están en regiones permitidas lo que valida la estructura modelada de la proteína Lack (21). Por otro lado, los resultados de alineamiento estructural con el programa PyMol (22), el modelo refinado de Lack con ModRefiner (7) y las estructuras de sus ortólogos reportadas en PDB son altamente similares (Tabla 1). Este modelo se convierte en una base importante para el desarrollo de moléculas que inhiban la función de Lack, ya que esta proteína está implicada en la infectividad y supervivencia del parásito (4). Estos mecanismos claves para el parásito se deben a que Lack interacciona con quinasas, fosfatasas, canales iónicos y, especialmente, con estructuras multiproteicas como el ribosoma (23-25). La estructura 3D modelada de Lack revela una estructura reportada de unión a ribosoma en Trypanosoma brucei conocida como RACK1 (PDB: 3zey) (25). La homología de Lack con la estructura de Rack1 muestra que aparentemente Lack podría jugar un papel importante regulando el comienzo de la síntesis de proteínas por medio del reclutamiento de la proteína quinasa C. Hasta ahora, los datos reportados demuestran que Lack y sus ortólogos son constituyentes de todos los ribosomas eucarióticos (23-26), a excepción del ribosoma de Trypanosoma cruzi (27), en donde no se observó la presencia de la proteína Rack1 en los ribosomas. De acuerdo a estos resultados, el modelo de Lack de L. (V.) panamensis supone la conservación de la función molecular (actividad biológica) en síntesis de proteínas en el parásito. Además, la proteína Lack participa en la conversión del plasminógeno en plasmina; esta interacción se ha postulado como una estrategia de internalización del parásito a la célula huésped (5).

Conclusiones
Nuestros resultados abren una ventana hacia la determinación por cristalografía de rayos-X de la proteína Lack. Estos hallazgos muestran la forma de obtener una proteína Lack plegada al suplementar el tampón de renaturalización con aditivos como L-arginina y β-ciclodextrina. El modelo estructural, demuestra que Lack es una proteína altamente conservada en los organismos eucarióticos.
Agradecimientos
Los autores agradecemos a Jairo Alonso Mesa Arango por su colaboración en la clonación del Lack. Agradecemos al CODI por la financiación del proyecto (IN1579CE).
Referencias
1. Alvar, J.; Vélez, I. D.; Bern, C.; Herrero, M.; Desjeux, P.; Cano, J.; Jannin, J.; de Boer, M. Leishmaniasis worldwide and global estimates of its incidence. PLoS ONE. 2012. 7: e35671. [ Links ]
2. Hide, M.; Marion, E.; Pomares, C.; Fisa, R.; Marty, P.; Bañuls, A. L. Parasitic genotypes appear to differ in leishmaniasis patients compared with asymptomatic related carriers. Int. J. Parasitol. 2013. 43: 389-397. [ Links ]
3. Peacock, C. S.; Seeger, K.; Harris, D.; Murphy, L.; Ruiz, J. C.; Quail, M. A.; Peters, N.; Adlem, E.; Tivey, A.; Aslett, M.; et al. Europe PMC Funders Group Comparative genomic analysis of three Leishmania species that cause diverse human disease. Nat. Genet. 2008. 39: 839-847. [ Links ]
4. Kelly, B. L.; Stetson, D. B.; Locksley, R. M. Leishmania major LACK antigen is required for efficient vertebrate parasitization. J. Exp. Med. 2003. 198: 1689-1698. [ Links ]
5. Gómez-Arreaza, A.; Acosta, H.; Barros-Álvarez, X.; Concepción, J. L.; Albericio, F.; Avilan, L. Leishmania mexicana: LACK (Leishmania homolog of receptors for activated C-kinase) is a plasminogen binding protein. Experimental Parasitology. 2011. 127: 752-761. [ Links ]
6. Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008. 9: 1-8. [ Links ]
7. Xu, D.; Zhang, Y. Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophysical Journal. 2011. 101: 2525-2534. [ Links ]
8. Karplus, K. SAM-T08, HMM-based protein structure prediction. Nucleic Acids Research. 2009. 37: W492-W497. [ Links ]
9. Kelley, L.; Sternberg, M. J. E. Protein structure prediction on the Web: a case study using the Phyre server. Nature Protocols. 2009. 4: 363-371. [ Links ]
10. Cudney, R.; Patel, S.; Weisgraber, K.; Newhouse, Y.; McPherson, A. Screening and optimization strategies for macromolecular crystal growth. Acta Crystallogr. D. Biol. Crystallogr. 1994. 50: 414-423. [ Links ]
11. Invernizzi, G.; Annoni, E.; Natalello, A.; Doglia, S. M.; Lotti, M. In vivo aggregation of bovine beta-lactoglobulin is affected by Cys at position 121. Protein Expression and Purification. 2008. 62: 111-115. [ Links ]
12. Tsumoto, K.; Umetsu, M.; Kumagai, I.; Ejima, D.; Arakawa, T. Solubilization of active green fluorescent protein from insoluble particles by guanidine and arginine. Biochemical and Biophysical Research Communications. 2003. 312: 1383-1386. [ Links ]
13. Tsumoto, K.; Umetsu, M.; Kumagai, I.; Ejima, D.; Philo, J. S.; Arakawa, T. Role of arginine in protein refolding, solubilization, and purification. Biotechnol. Prog. 2004. 20: 1301-1308. [ Links ]
14. Vallejo, L. F.; Rinas, U. Strategies for the recovery of active proteins through refolding of bacterial inclusion body proteins. Microbial Cell Factories. 2004. 3: 11. [ Links ]
15. Oganesyan, N.; Kim, S.; Kim, R. On-column Chemical Refolding of Proteins. Pharma Genomics. 2004. September: 22-26. [ Links ]
16. Begg, G. E.; Speicher, D. W. Mass Spectrometry Detection and Reduction of Disulfide Adducts Between Reducing Agents and Recombinant Proteins with Highly Reactive Cysteines. Journal of Biomolecular Techniques. 1999. 10: 17-20. [ Links ]
17. McPherson, A. Introduction to protein crystallization. Methods. 2004. 34: 254-265. [ Links ]
18. Kim, Y.; Babnigg, G.; Jedrzejczak, R.; Eschenfeldt, W. H.; Li, H.; Maltseva, N.; Hatzos-Skintges, C.; Gu, M.; Makowska-Grzyska, M.; Wu, R.; et al. High-throughput protein purification and quality assessment for crystallization. Methods. 2011. 55: 12-28. [ Links ]
19. Preckel Tobias, V. M. and K. M. Quality Control for the Agilent 2100 Bioanalyzer Protein 200 Plus LabChip Kits. Technical Overview; 2002. 1-8. [ Links ]
20. Saridakis, E.; Dierks, K.; Moreno, A.; Dieckmann, M. W. M.; Chayen, N. E. Separating nucleation and growth in protein crystallization using dynamic light scattering. Acta Crystallographica Section D Biological Crystallography. 2002. 58: 1597-1600. [ Links ]
21. Lovell, S. C.; Davis, I. W.; Arendall, W. B.; de Bakker, P. I. W.; Word, J. M.; Prisant, M. G.; Richardson, J. S.; Richardson, D. C. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins. 2003. 50: 437-450. [ Links ]
22. Baugh, E. H.; Lyskov, S.; Weitzner, B. D.; Gray, J. J. Real-time PyMOL visualization for Rosetta and PyRosetta. PLoS ONE. 2011. 6: e21931. [ Links ]
23. Ullah, H.; Scappini, E. L.; Moon, A. F.; Williams, L. V.; Armstrong, D. L. E. E. Structure of a signal transduction regulator, RACK1, from Arabidopsis thaliana. Protein Science. 2008. 17: 1771-1780. [ Links ]
24. Coyle, S. M.; Gilbert, W. V; Doudna, J. Direct link between RACK1 function and localization at the ribosome in vivo. Molecular and cellular biology. 2009. 29: 1626-1634. [ Links ]
25. Hashem, Y.; des Georges, A.; Fu, J.; Buss, S. N.; Jossinet, F.; Jobe, A.; Zhang, Q.; Liao, H. Y.; Grassucci, R.; Bajaj, C.; et al. High-resolution cryo-electron microscopy structure of the Trypanosoma brucei ribosome. Nature. 2013. 494: 385-389. [ Links ]
26. Sengupta, J.; Nilsson, J.; Gursky, R.; Spahn, C. M. T.; Nissen, P.; Frank, J. Identification of the versatile scaffold protein RACK1 on the eukaryotic ribosome by cryo-EM. Nature structural & molecular biology. 2004. 11: 957-962. [ Links ]
27. Gao, H.; Ayub, M. J.; Levin, M. J.; Frank, J. The structure of the 80S ribosome from Trypanosoma cruzi reveals unique rRNA components. Proc Natl Acad Sci. 2005. 102: 10206-10211. [ Links ]
