











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
Plasmodium falciparum es un parásito protozoário intracelular causante de la forma más grave de malaria, una enfermedad de la cual en el último año se reportaron cerca de 212 millones de casos nuevos y 429.000 muertes 1. Dado que en la actualidad no existe una vacuna de uso clínico y ante el aumento de la resistencia del parásito contra medicamentos antimaláricos (e.g. cloroquina, artemisinina, etc.), la Organización Mundial de la Salud, en colaboración con diferentes laboratorios del mundo, se encuentra en la búsqueda de proteínas que sirvan como blancos terapéuticos para el control de la enfermedad 2.
En los últimos 20 años se ha venido estudiando la nicotinamida/ nicotinato mononucleótido adenililtransferasa NMNAT (EC 2.7.7.1/18), enzima en la cual convergen las rutas de novo y de reciclaje del dinucleótido de adenina y nicotinamida NAD(H) 3. El NAD(H) y su derivado fosforilado NADP(H) son esenciales en el metabolismo celular y, en el caso de P. falciparum, en el proceso de infección eritrocítica 4 - 6 ). Durante su ciclo de vida asexual, el parásito infecta a los glóbulos rojos y degrada la hemoglobina, tras lo cual incrementa el estrés oxidativo. Este aumento activa los sistemas antioxidantes del parásito que dependen del poder reductor del NADP(H) celular. Así mismo, en el proceso de infección, las rutas metabólicas, como el ciclo de las pentosas fosfato y la glicólisis, aumentan su actividad. Estas rutas dependen de NADP+ y NAD+, respectivamente 7 , 8 ).
La NMNAT de P. falciparum ha sido identificada y estudiada enzimáticamente como proteína recombinante en trabajos anteriores 9 - 11 ). Durante su expresión, se producen grandes cantidades de agregados insolubles conocidos como cuerpos de inclusión (Cl o IB, acrónimo en inglés), compuestos principalmente por proteína mal plegada de poca utilidad respecto a la proteína soluble y de dispendiosa purificación. Con el fin de maximizar la cantidad de proteína recombinante soluble, se han propuesto varias estrategias: cambios en las condiciones de expresión, cepas, vectores, promotores y etiquetas 12 ). En este trabajo se reportan los resultados de la evaluación de varios vectores y etiquetas en la expresión y solubilidad de la NMNAT recombinante de P. falciparum (PfNMNAT).
Materiales y métodos
Estimación in silico de la solubilidad
Se escogieron los cuatro vectores más promisorios en cuanto a solubilidad, teniendo en cuenta tres aspectos: expresión, solubilidad y purificación 13 ). Los cuatro vectores escogidos fueron: Champion™ pET SUMO (ThermoFisher), el cual le otorga la etiqueta de 6His+SUMO (His + SUMO); pCold TF DNA (Takara), que fusiona la etiqueta de 6His+Chaperona Trigger Factor (His + TF); pBAD202/D-TOPO (Invitrogen), fusiona la etiqueta de Tiorredoxina (Trx) y pMAL-c5X (New England Biolabs) que su proteína fusión es la proteína de unión a maltosa (MBP).
Se implementaron tres programas bioinformáticos, SOLPro 14 ), PROSO II 15 y ESPRESSO 16, que estiman la solubilidad de la proteína recombinante expresada a partir del sistema heterólogo E. coli. Para esto, se tomó la secuencia primaria de la PfNMNAT (PF13_0159), unida a las diferentes etiquetas moleculares aportadas por los vectores de expresión, y se identificó la probabilidad de encontrar esta proteína en la fracción soluble.
Amplificación de la región codificante de la PfNMNAT
Para la amplificación de la región codificante de la PfNMNAT se usaron varias parejas de iniciadores que permitieron la clonación en los respectivos vectores de expresión (Tabla 1). Las condiciones utilizadas para la amplificación fueron: 1 U de la enzima Taq Polimerasa (Applied Biosystems) o Pfu DNA Polimerasa (ThermoFisher), MgCl2 o MgSO4 (2,5 mM), buffer de la ADN polimerasa (1X), dNTP's (10 mM), H2O DEPC, iniciadores y aproximadamente 50 ng de ADN plantilla; todo a un volumen final de 15 μL. Se empleó el siguiente perfil térmico: un ciclo de desnaturalización inicial a 94 °C durante 10 min, seguido por 30 ciclos de 94 °C por 45 s, 50 °C por 1 min, 72 °C por 45 s, y una extensión final a 72 °C por 10 min (termociclador Biorad) 10.

Clonación de la región codificante de la PfNMNAT
Los productos de PCR fueron visualizados en geles de agarosa 1%. Para realizar la clonación se usaron dos estrategias dependiendo del vector de expresión: ligación directa con el producto de PCR o subclonación utilizando el vector pGEM®-T Easy (PROMEGA). La ligación se realizó teniendo en cuenta una relación 1:1 inserto purificado y vector (aproximadamente 50 ng).
Transformación en cepas de E. coli
Se transformaron cepas de E. coli de mantenimiento One Shot Mach1™ T1 (ThermoFischer) y de expresión BL21(DE3) (Sigma-Aldrich) químicamente competentes, mediante choque térmico (42 ° C durante 45 s, 5 min en hielo). Inmediatamente, las bacterias transformadas se incubaron durante 1 h a 37 °C en medio SOC (triptona 2%, extracto de levadura 0,5%, NaCl 10 mM, KCl 2,5 mM, MgCl2 10 mM y glucosa 20 mM). Luego, se sembraron en cajas de Petri con LB-agar y antibiótico de selección respectivo para cada vector.
Purificación del ADN recombinante
Se partió de un cultivo de 10 mL, inoculando en medio LB líquido suplementado con el antibiótico de selección. Se utilizó el método de lisis alcalina 17 ).
Expresión de las proteínas recombinantes
Se tomó una colonia de cada uno de los plásmidos recombinantes y se inocularon individualmente en medio LB, suplementado con el antibiótico de selección; se dejaron creciendo toda la noche a 37 °C. Se realizó una dilución del cultivo 1:100 y se continuó el crecimiento hasta alcanzar una D.O - 0,6 a 600 nm. Se realizó la inducción de la expresión de las proteínas siguiendo las recomendaciones del fabricante con IPTG (Isopropil β-D-1-tiogalactopiranósido) o arabinosa, según el caso, incubando a 15-25 °C con agitación constante. Finalizada la inducción, se colectaron las bacterias por centrifugación a 6000 rpm por 15 min a 4 °C; se descartó el sobrenadante y se determinó el peso húmedo de células.
Lisis celular
Los pellets celulares se resuspendieron en buffer de lisis (5 mL/g). Se adicionó lisozima (Sigma-Aldrich) a una concentración final de 1 mg/ mL; ARNasa (ThermoFisher), a una concentración final de 6,25 μg/ mL, y cocktail de inhibidores de proteasas (Sigma-Aldrich) P8340; AEBSF 1 mM, E64 14 μM, Pepstatin A 15 μM, Bestatin 40 μM, Leupeptin 20 μM, Aprotinin 0,8 μM). El conjunto se incubó en hielo durante 30 min con agitación mecánica. Pasado este tiempo, se sometió la muestra a 5 min de sonicación sobre hielo (50% de amplitud, 15 s de pulso y 15 s de reposo). La muestra fue centrifugada a 12000 rpm por 20 min a 4 °C, separándose el sobrenadante (fracción soluble) y el pellet (fracción insoluble o cuerpos de inclusión).
Detección de las proteínas recombinantes por electroforesis SDS-PAGE y western blot
Los lisados de los clones inducidos se analizaron por electroforesis vertical SDS-PAGE. Para esto se tomó una alícuota de las fracciones solubles obtenidas en la lisis y se resuspendieron en buffer de carga desnaturalizante. Las muestras se calentaron a 92 °C durante 7 min. La visualización de las bandas se logró mediante la tinción del gel con azul de Coomassie. Para los ensayos de inmunodetección (western blot), las proteínas separadas por SDS-PAGE fueron transferidas a membrana de nitrocelulosa (ThermoFisher), usando el método de electrotransferencia húmeda 18 ), en buffer de transferencia aplicando 200 mA por 2 h. A su término, se tiñó la membrana con Rojo Ponceau (Sigma-Aldrich) y se realizó la inmunodetección. Se identificó el sistema de expresión que tuvo mayor aumento de solubilidad mediante la confirmación en western blot y se realizaron ensayos de purificación.
Purificación de la proteína recombinante por afinidad a resina de amilosa
La proteína MBP-PfNMNAT (MBP, maltose-binding protein) se purificó a partir de la fracción soluble mediante afinidad a resina de amilosa (NEB). La resina se equilibró con buffer de lisis y se mezcló con la fracción soluble diluida (1:6). La unión se llevó a cabo durante 1 h a 4 °C con agitación constante. La mezcla se transfirió a una mini columna, empacándose la resina y obteniéndose las proteínas no unidas. La resina se lavó con 12 volúmenes de buffer de lisis.
La proteína MBP-PfNMNAT se eluyó con buffer de elución (buffer de lisis + maltosa 10 mM); se recolectaron 25 fracciones de aprox. 60 μL, que fueron suplementadas con glicerol (10% v/v) y almacenadas a -80 °C. La proteína en la fracción soluble y en los eluídos se cuantificó mediante el método de Bradford 19.
Ensayo de actividad enzimática
Se realizó un ensayo in vitro para determinar la actividad de la proteína recombinante purificada. Las mezclas de reacción (mononucleótido de nicotinamida (NMN) 1 mM y ATP 1,3 mM en buffer HEPES 100 mM + Mg2+ 10 mM, pH 7,5) se incubaron a 37 °C durante 30 min, al término de lo cual se detuvo la reacción con HClO4 1,2 M a 4 °C. La proteína se precipitó mediante centrifugación a 12000 rpm durante 3 min a 4 °C. Se neutralizó el sobrenadante con K2CO3 1 M en hielo. El resultado de la reacción in vitro se evaluó mediante HPLC en fase reversa. Se utilizó una columna Phenomenex Luna C18 (250 mm x 4,60 mm; 5 μm). El volumen de inyección fue de 50 μL. Fases móviles: buffer fosfato de potasio 0,12 M pH 6,0 y metanol. Flujo constante de 1,5 mL/min, con un tiempo de 20 min por corrida a temperatura ambiente. Detección espectrofotométricamente a 254 nm.
Resultados y discusión
Estimación in silico de la solubilidad
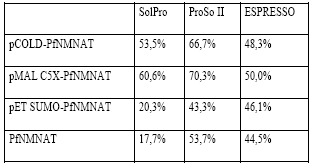
El estudio bioinformático indicó una mayor solubilidad de la proteína PfNMNAT expresada desde el vector pMAL-c5X. Los resultados para la PfNMNAT expresada desde los otros vectores no fueron concluyentes, pues se encontraron divergencias con los programas empleados. Aunque todos los programas predicen solubilidad del 50% aproximadamente, como control se usa la secuencia primaria de la proteína sin ninguna etiqueta, donde se puede observar que la expresión de esta proteína en el sistema heterólogo sería insoluble.
Lo anterior es predecible, debido a que es una proteína externa de la bacteria y por ende no se puede resolver su plegamiento. Por tanto, es importante adicionar las etiquetas para obtener la versión recombinante de forma soluble (Tabla 2). Entonces, se evaluó el desempeño de estos vectores y etiquetas en la solubilidad experimentalmente.
Tabla 2 Predicción in silico de la solubilidad de las proteínas recombinantes PfNMNAT usando programas bioinformáticos
Amplificación, clonación y transformación
Se obtuvieron los productos de PCR deseados para cada pareja de iniciadores. La clonación en los tres vectores empleados fue exitosa, después del análisis por PCR y digestión con enzimas de restricción (Figura 1).

Figura 1 Verificación de los plásmidos recombinantes de la PfNMNAT mediante PCR. 1: Marcador de peso AxyGen 100 pb, 2 y 3: ADN plasmídico extraído pCOLD-PfNMNAT, 4 y 5: ADN plasmídico extraído pMal c5X-PfNMNAT 6: Control positivo de la reacción y 7: Negativo de la reacción. (Tamaño esperado 615 pb). Gel de agarosa 1% teñido con bromuro de etidio
Expresión de las proteínas recombinantes
En la figura 2 se muestra el perfil electroforético de cada uno de los sistemas de expresión evaluados. Se observa una mayor expresión de la proteína con la etiqueta MBP ( ~ 67,3 kDa) respecto a las otras etiquetas: His + SUMO ( ~ 5,8 kDa), Trx ( ~ 37,8 kDa) y His-TF (76,8 kDa), tal y como se predijo en los programas bioinformáticos. Hasta el día de hoy no se tiene claro el mecanismo por el cual la proteína fusión puede modificar la solubilidad de la proteína de interés, sin embargo, la MBP es una de las más estudiadas. La proteína de unión a maltosa puede estar funcionando como una chaperona molecular que, mediante su bolsillo hidrofóbico, secuestra la proteína con plegamiento intermedio y le da una segunda oportunidad de plegamiento. Esto permite que la proteína recombinante tenga el plegamiento nativo y, por ende, se encuentre en la fracción soluble; si, por el contrario, no se puede resolver el plegamiento, formará agregados insolubles 20 , 21.

Figura 2 Análisis de la expresión soluble de las proteínas recombinantes PfNMNAT en BL21 (DE3). 1. Patrón de peso molecular en kDa (ThermoFisher). 2. Proteínas solubles BL21 (DE3) sin transformar. 3. Proteínas solubles expresión de His o SUMO-PfNMNAT. 4. Proteínas solubles expresión de Trx-PfNMNAT. 5. Proteínas solubles expresión de MBP-PfNMNAT. 6. Proteínas solubles expresión de His o TF-PfNMNAT. A. SDS-PAGE 12% teñido con azul de Coomassie. B. Inmunodetección en membrana de PVDF. (Las flechas indican las proteínas sobreexpresadas y reconocidas en el western blot, las bandas inespecíficas son comunes hasta en el control negativo y se deben a reconocimiento con el anticuerpo primario)
La proteína MBP fusionada a la PfNMNAT en la región amino terminal permite realizar la purificación de la proteína recombinante mediante cromatografía de afinidad, debido a la elevada interacción de la resina de amilosa por las proteínas recombinantes que contienen la etiqueta. MBP consta de dos dominios estructurales en forma de bisagra que le permiten interactuar con diversos azúcares como la amilosa y la maltosa. La unión con ellas genera un cambio conformacional que cierra la proteína con la cual se enlaza al azúcar a través de enlaces de puentes de hidrógeno y por contactos de Van der Waals, principalmente con residuos aromáticos, causando un apilamiento de las cadenas laterales contra la cara de los anillos de azúcar.
Se espera que el tamaño de la proteína fusión (42,5 kDa) no interfiera en la estructura y función de la proteína purificada, lo cual se confirmó mediante ensayos enzimáticos posteriores.
Purificación mediante afinidad a amilosa
La purificación de la proteína se llevó a cabo en condiciones nativas, buscando no interferir en la funcionalidad y estructura. La purificación por cromatografía de afinidad se siguió mediante SDS-PAGE (figura 3).

Figura 3 Expresión y purificación de la MBP-PfNMNAT. 1. Patrón de peso molecular en kDa (ThermoFisher), 2. Proteínas totales expresión etiqueta MBP (Control), 3. Proteínas totales expresión de MBP-PfNMNAT, 4. Proteínas solubles expresión de 15 MBP-PfNMNAT, 5. Eluído de la purificación de la MBP-PfNMNAT. SDS-PAGE 10% Teñido con azul de coomassie. * Corresponde a MBP como producto de degradación
Se observó que, a partir de los lavados, se eliminó una gran cantidad de proteínas no unidas, permitiendo obtener eluídos con la proteína recombinante parcialmente pura y concentrada. Se presentó una pequeña contaminación por una proteína de aproximadamente 45 kDa correspondiente a un producto de degradación, debido a una alta tasa de degradación de la proteína recombinante PfNMNAT sobreexpresada, y el tamaño corresponde a la etiqueta (MBP) que por el contrario es muy estable.
Esta información se corroboró mediante western blot usando anti-MBP, en el cual se observó reconocimiento del producto de degradación (Figura 4).

Figura 4 Purificación de la proteína recombinante MBP-PfNMNAT. 1: Patrón de peso molecular en kDa (ThermoFisher), 2. Fracción soluble, 3: Purificación de MBP-PfNMNAT. A. SDS-PAGE 12% teñido con azul de coomassie, B. Inmunodetección en membrana de PVDF. Anticuerpo 1°: Anti-MBP 1:10000. *producto de degradación MBP
Ensayos de actividad enzimática
La actividad nucleotidiltransferasa de la proteína recombinante se evaluó mediante ensayos NMNAT directos y la posterior observación del pico correspondiente al NAD+ sintetizado por HPLC en fase reversa (Figura 5). Respecto a la actividad encontrada en la fracción soluble, en el eluído de la purificación por afinidad fue 18,4 veces mayor (Tabla 3), permitiendo obtener buenos rendimientos para la proteína recombinante.
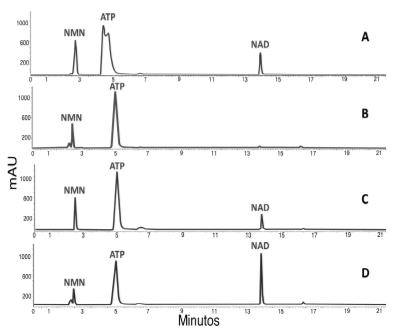
Figura 5 Verificación de la actividad catalítica de la MBP-PfNMNAT cuantificada en RP-HPLC. A. Patrones, B. Control negativo (Buffer de elución de la proteína), C. Fracción soluble. D. MBP-PfNMNAT

Conclusiones
La fusión de la etiqueta MBP permitió un aumento en la solubilidad de la proteína PfNMNAT en comparación con las otras etiquetas usadas, lo cual, además, facilitó la obtención de la proteína con un alto porcentaje de purificación y buen rendimiento. Sin embargo, la adición de etiquetas de solubilización para la producción de proteínas recombinantes no garantiza la solubilidad de la proteína a expresar, ya que depende de características intrínsecas de cada proteína y de la interacción con la etiqueta.
Se recomienda el uso de programas bioinformáticos como la primera aproximación para el uso de las etiquetas, con el objetivo de determinar su posible efecto sobre la solubilidad. No obstante, hay que tener precaución con los resultados de la predicción, ya que estos dependen de la matriz de proteínas de comparación.

