











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroduction
A wide diversity of microbial species interacts in natural ecosystems, contributing to waste degradation (Sun et al., 2013; Azman et al., 2015; Raja et al., 2017). The use of agricultural crop residues and agro-industrial waste would be attractive alternatives as a source of renewable energy because they close production cycles in a circular economy approach. However, despite the diversity of microorganisms involved in the decomposition of organic matter, the efficient biodegradation of these residues remains a challenge for bioethanol and biogas production due to the high percentages of lignocellulosic material, whose polymers are difficult to decompose (Cortés, 2016; Widjaja et al., 2016; Amin et al., 2017). The search continues, therefore, for microorganisms that naturally contain this lignocellulolytic capacity that can be exploited biotechnologically.
Teuro Higa developed in 1986 the technology of so-called efficient microorganisms (EM) (Allahverdiyev, Kirdar et al., 2011; Higa & Parr, 2013) that are isolated from undisturbed soils and ecosystems. EM technology has been applied in agriculture and forestry in more than 80 countries worldwide (Calero Hurtado, Pérez Díaz et al., 2019; Tanya & Leiva-Mora, 2019) with beneficial impacts. Microbial community compositions related to anaerobic EM consortia have not been characterized, but previous studies have identified phylogenetic groups, including Actinomycetales and fermenting fungi, photosynthetic bacteria, yeasts, and lactic acid bacteria (Namasivayam et al., 2014; Joshi et al., 2019).
Soil microorganisms, including EM consortia, play a crucial role in agricultural production and environmental decontamination, as they allow the availability of assimilable nutrients for plants during the biodegradation of organic matter, where phytohormones and secondary metabolites are released (López-Dávila, Gil Unday et al., 2017; Calero Hurtado, Quintero Rodríguez et al., 2019; Joshi et al., 2019; Tanya & Leiva-Mora, 2019; Castro et al., 2022). However, physico-chemical characteristics and microbiological composition of EM consortia should be clarified in order to understand their beneficial effects on agricultural production (Calero Hurtado et al., 2020). The beneficial uses of EM consortia include restoration of damaged soils after application of chemical fertilizers and pesticides (Alvarez et al., 2018), manufacturing of fermented fertilizers (biofertilizers) (Calero Hurtado, Quintero Rodríguez et al., 2019), processing of organic waste, and wastewater treatment (Allahverdiyev, Atilla et al., 2011). It has been widely reported that Lactobacillus species are prevalent in the EM consortia that is the basis for their use as a biofertilizer to enhance agricultural production (Blainski et al., 2018; Daranas et al., 2018; Quattrini et al., 2018; Naik et al., 2019; Abd El-Mageed et al., 2020; Muhialdin et al., 2020).
Therefore, an important strategy would be to determine not only the main physicochemical characteristics but also which of them contribute to the improvement of soil properties, growth of plants, and other important environmental processes (Higa & Parr, 2013). The EM consortia works synergistically to release beneficial substances such as vitamins, hormones, enzymes, organic acids, bioactive minerals, and various antioxidants when they come into contact with the soil organic matter (Allahverdiyev, Kirdar et al., 2011). In addition, the EM consortia helped to improve the soil pH and increase the action of mineral nutrients, hormones, and other metabolites that accelerate decomposition of organic wastes and increase formation of biogas (López-Dávila, Gil Unday et al., 2017). Therefore, the hydrolytic and fermentative activities of the EM consortia could be potentially used as a pre-treatment or biostimulant in the agricultural waste bioconversion processes into bioenergy, for instance in bioethanol or biogas production.
The microbial dynamics of natural ecosystems (soils) as well as artificial ecosystems (anaerobic reactors), can be very complex due to the wide diversity of species and metabolic interactions required to degrade the organic matter. Microbiological characterization of these ecosystems using culture-dependent techniques are insufficient since only 0.1-10% of the bacteria in the environment can be cultivated (Nobu et al., 2015; Saw et al., 2015; Jiménez-Hernández et al., 2021). Hence, it is necessary to apply molecular methods that allow the analysis of taxonomic diversity and spatial structure of complex microbial communities to identify specific microbial populations in their natural habitat as well as to predict existing metabolic interactions.
Previous studies for microbiome monitoring employed molecular techniques based on 16S rRNA gene analysis such as denaturing gradient gel electrophoresis (DGGE), terminal restriction fragment length polymorphisms (TRFLP), single-strand conformation polymorphism (SSCP) or capillary sequencing of selected genes by the Sanger method (Zoetendal et al., 2008). In complex microbial ecosystems, these methods provide an incomplete composition of the microbial community, showing only the existence of certain groups. The development of metagenomic approaches based on next-generation sequencing (NGS) has been one of the great advances in molecular biology (Zhou et al., 2015; Sanz & Kõchling, 2019). The combination of different technologies seems to be imperative to reach the goals. Techniques complement each other, being the weaknesses of some and the strengths of others, especially when the study requires the identification of species to elucidate predominant metabolic pathways (Carabeo-Pérez et al., 2019).
Since 2014, in the Microbiology Laboratory of the University of Sancti Spíritus (Universidad de Sancti Spíritus "José Martí Pérez" - UNISS, Cuba), EM consortia have been developed, taking into account the methodology proposed by Teuro Higa and modified by Olivera-Viciedo et al. (2014). These studies were focused on the application of EM as a biostimulant to soils and plants. However, a detailed characterization of this microbial consortium has not been performed.
Agricultural soils in Cuba are currently in unfavorable conditions due to intensive cultivation and inadequate management (Febles-González et al., 2014). Most of the agricultural soils in this overexploited condition are located in the central region of the country that is a major producer of sugarcane (ONEI, 2020). These are brown carbonate soils with a pH between 5.0 and 8.5. In addition, they have high N, P and K content due to fertirrigation with wastewater from sugar factories and distilleries (Crespo et al., 2018). However, the diversity and dynamics of the microbial populations present in this soil and how they contribute to the degradation of agricultural residues were not found in previous studies.
There are also extensive areas of soil in Cuba that have very little anthropogenic action and are of special interest. These are generally forested areas covered by bamboo species (Phyllostachys reticulata [Ruprecht] Koch) and marabu (Dichrostachys cinerea [L.] Wight & Arnott). In the case of bamboo, they are generally found in forested areas near rivers or canals with alluvial soils (non-carbonate) (Oca-Risco et al., 2014), and marabu species can germinate in any type of soil. No further molecular characterization of these virgin ecosystems was done in this study.
The objective of this research was to characterize the microorganisms isolated from two soil communities (sugar cane crop and bamboo forest) and the efficient microorganisms obtained in the laboratory (EM16 consortium), taking into account physico-chemical characteristics, diversity, quantification, and taxonomic identification through molecular techniques.
Materials and methods
Obtaining isolates of EM consortium and agricultural soils
The EM consortium was produced in the Microbiology Laboratory of the University of Sancti Spíritus (UNISS, Cuba) pilot plant (20 L fermenter), following a methodology similar to that proposed by Olivera-Viciedo et al. (2014).
Production was carried out as follows: 5 L of whey was mixed with 5 L of "C molasses" from a sugarcane factory. This mixture was added in layers to 10 kg of decomposing foliage and soil obtained from virgin forests undisturbed by human activity in the vicinities of the Zaza River of the Southern region of Sancti Spiritus province. Nine kg of corn flour was added to the mixture which was homogenized, compacted, and sealed in an anaerobic tank. The mixture (solid EM material) was kept at 28°C in the dark for 21-25 d for fermentation. After this time, 2 kg of this solid EM material was diluted in 18 L of distilled water containing 1 L of whey and 1 L of "C molasses" to obtain a total volume of 20 L of liquid bioproduct. The culture was kept hermetically sealed for 7 d under the same conditions described above, to avoid disturbing the fermentation process. The consortium obtained was called EM16.
To obtain the culture of soil microorganisms, 10 samples of 20 g were collected from two different agroecosystems: i) 10 cm of soil beneath the leaf floor of sugarcane field 7 d after harvest and ii) 10 cm of soil beneath the leaf floor of 5-year-old bamboo (Bambusa vulgaris Schrader) field (brown soils with carbonates and alluvial). Each soil sample was individually suspended in 350 ml of peptone cellulose solution (PCS medium: 0.1% yeast extract, 0.5% peptone, 0.2% CaCO3, 0.5% NaCl, 0.5% cellulose, pH 7.0) for 7 d at 30°C in static culture flasks to isolate microbial strains capable of degrading lignocellulosic substrates.
Physico-chemical characterization of EM
The EM16 consortium and the soil isolates grown for 7 d in PCS were characterized by the following physico-chemical parameters: dry matter (DM), volatile solids (VS), ash, and pH, according to the 23rd Standard Methods for the Examination of Water and Wastewater (Baird et al., 2017).
Morphological analysis (growth in plates)
To identify the cultural bacteria, NA (nutrient agar) medium was used, employing surface spreading plate technique, with 0.1 ml of original samples and dilutions 10-1, 10-2 and 10-3 as inoculum. Petri plates were incubated at 37°C for 96 h, after which it was possible to differentiate the colonies by their morphological characteristics in the culture.
DNA extraction
Genomic DNA was extracted from each sample (triplicate aliquots of 200 mg) using the FastDNA® SPIN Kit for Soil and the FastPrep® Instrument (MP Biomedicals, USA) according to the manufacturer's guidelines. The quantity and purity of DNA were determined photometrically using a NanoDrop 2000/2000c (Thermo Scientific, USA) according to the manufacturer's guidelines. Isolated DNA was stored at -20°C until further processing.
Amplification of bacterial 16S rRNA gene sequences
Genes encoding for bacterial 16S rRNA (rrs) were amplified using the PCR primers 27f(5'-AGAGTTTGATCMTGGCT-CAG-3') (Lane, 1991; Sipos et al, 2007) and 1492r (5'- TAC-GGYTACCTTGTTACGACTT-3') (Weisburg et al., 1991; Després et al., 2007). The PCR reaction mix contained 1X Taq buffer, 2 mM MgCl2, 0.2 mM of each deoxynucleoside triphosphate, 0.4 of each primer, 1 (10 ng approximately) of template DNA and 1 U of Taq DNA polymerase for a final volume of 25 μl. The amplification was performed with an initial denaturation step for 1 min at 94°C, followed by 34 cycles of denaturation at 94°C for 1 min, annealing at 57°C for 1 min, extension at 72°C for 2 min and a final extension stage at 72°C for 3 min.
Chemicals and enzymes were provided by Fermentas (St. Leon-Rot, Germany), Promega (USA) Corporation and Molecular Biology (Thermo Scientific, USA). The correct length of the amplicons was verified by 1% agarose gel electrophoresis, using the GeneRuler® 1kb Plus DNA Ladder (Thermo Scientific, USA). To minimize the risk of any PCR bias, three parallel PCR reactions were performed for each of the three parallel DNA extracts obtained from each environmental sample. The concentration of the amplified 16S rRNA gene fragments was measured using the Qubit4® fluorometer (Invitrogen, Thermo Scientific, USA).
Terminal Restriction Fragment Length Polymorphism analysis (TRFLP)
For the bacteria diversity study, TRFLP analysis was carried out following the protocol proposed before by Rademacher et al. (2012). The primers 27f (5'-AGAGTTTGATCMT- GGCTCAG-3') (Lane, 1991; Sipos et al., 2007), labeled at the 5' terminal end with Indodicarbocianin (Cy5) and 926r (5'-CCGTCAATTCMTTTRAGTTT-3') (Weisburg et al., 1991; Després et al., 2007) were used. The three independent amplification products, based on the same DNA template, were pooled and purified by applying the PureLink® PCR Purification Kit (Invitrogen, Thermo Scientific, USA). The concentration of purified products was measured using the Qubit4® fluorometer (Invitrogen, Thermo Scientific, USA).
After purification, the PCR products were digested with MspI and Hin6I following the manufacturer guidelines for each restriction enzyme. The digestion fragments were electrophoretically separated and detected by fluorescence using a GenomeLab® GeXP Genetic Analysis System (Beckman Coulter, Krefeld, Germany). The data obtained were analyzed using the DataConnect® software (Applied Biosystems interface-Thermo Fisher) considering the size calculation of the detected terminal restriction fragments (TRFs) based on the migration time of the applied size standard. All fragments with a sequence length between 60 and 640 bp were used for further analyses with the T-Rex software package, available online (http://trex.biohpc.org). The identification of "true" peaks by distinguishing baseline "noise" from signals of fluorescently labeled fragments, as well as the alignment of TRFs with a threshold of 0.5, was based on the evaluation of the peak height. In the last evaluation step, TRFs were visualized by their relative distribution, considering that TRFs with a relative abundance lower than 2% were removed from the analyses.
Bacterial 16S RNAr gene sequencing by Ion Torrent
To amplify the 16S hypervariable regions, the Ion 16S® Metagenomics Kit (Life Technologies, Thermo Scientific, USA) was used, on the Ion Torrent Personal Genome Machine® (PGM, Ion Torrent) platform, following the manufacturer guidelines. The remaining PCR reaction was purified using the PureLink PCR Purification Kit (Life Technologies, Thermo Scientific), and the concentration of the purified amplicons was assessed with the Qubit4 fluorometer and Qubit® dsDNA High Sensitivity Assay Kit (Life Technologies, Thermo Scientific). The DNA library construction, quantification, template preparation, and sequencing were developed according to the standard protocol, well-described by Adamiak et al. (2018). Metage-nomic data was evaluated using the Ion Reporter® software (Invitrogen, Thermo Scientific, USA), showing the taxonomic distribution of the target microbial community, based on the software database. The raw data of sequences have been deposited in the Sequence Read Archive (SRA) of the National Center for Biotechnology Information (NCBI) under the Bioproject accession number PRJNA764521 (http://www.ncbi.nlm.nih.gov/bioproject/764521).
Analysis of the diversity and organization of the microbial communities
The bacteria richness (Rr) considering more than 1% of abundance was calculated by the methodology proposed by Marzorati et al. (2008), based on the number of OTUs obtained in each electropherogram or cluster. The Shannon-Weaver diversity index (H) (Shannon & Weaver, 1963) was used to evaluate and compare this diversity, and the evenness index (Pielou index, H/Hmax) of the microbial community in each sample was calculated as the quotient of the Shannon index and the potential number of species in the sample (Hmax). This index describes the uniformity of the distribution of individuals in the community (Pielou, 1966).
Results and discussion
Physico-chemical characterization of EM16
The EM16 consortium presented low contents of dry matter (DM), which ranged from 0.90% to 1.57% for fresh matter (FM). Also, more than 50% of DM are volatile solids (VS) (0.51-0.95 of VS (% FM)), and 56.8-60.67 VS (% DM) is attributable to the biomass content. During the 7 d anaerobic fermentation process, biomass production is limited due to the anaerobic condition (Madigan et al., 2019). In addition, the dilution during the EM preparation described above (EM and agricultural soils isolates obtention) could contribute to this measure. Dai et al. (2016) also described a low organic matter content in EM bioproducts. The EM16 consortium had acid properties (pH = 4.31-4.46), agreeing with other reports where the pH ranged from 3.1 to 4.7 (López-Dávila, Calero Hurtado et al., 2017; Calero Hurtado et al., 2020). In the EM16 consortium, the low pH might be due to: i) the use of substrates with low pH like whey and molasses (Núñez-Caraballo et al, 2019; González-Herrera et al., 2021) and ii) the volatile organic acids formed during the fermentation processes (Xiong et al., 2012; Dai et al., 2016). In addition, the low pH reported by Calero et al. (2020) could be linked to the assimilation of some important mineral nutrients, such as N, P, K, and Ca that help to improve soil properties and plant growth. Also, knowledge of this characterization could help to identify substances important for use in agricultural production and other environmental practices.
Morphological analysis
The EM16 sample did not showed colony formation on plate growth (in aerobic condition). This result may be related to the lower relative abundance of the bacterial species in this sample with respect to the other isolated soil or that these microorganisms were not capable of growing under these conditions. EM consortium is frequently composed mainly of Lactobacillus species (Tanya & Leiva-Mora, 2019) that are facultative anaerobic bacteria difficult to grow in a Petri dish. On this basis, Calero et al. (2020) reported that the EM consortium is composed of some species of Lactobacillus bulgaricum, Bacillus subtilis and Saccharomyces cerevisiae that represent microbial diversity. Therefore, in the EM16 consortium, some of these species should also be identified.
In contrast, in the undiluted soil isolate samples, bacterial growth exceeded 300 colonies after 24 h of incubation; those plates are reported as uncountable. However, the diluted samples (10-2 and 10-3) showed colony growth. After the incubation period, it was possible to differentiate the bacteria colonies according to the different morphological characteristics in the culture.
In sugarcane soil isolates, bacterial colonies were observed mostly in a circular, irregular, or rhizoid shape, with rounded, wavy, or lobed edges and different surfaces such as flat, convex, accumulated, and umbilical. Bamboo soil isolated colonies with irregular or circular shapes with flat surfaces and wavy edges predominated.
The observed morphological diversity demonstrated that a great variety of metabolic pathways may also exist in soil microorganisms. This physiological diversity allows the microbial community to maintain the balance of agroecosystems. To guarantee soil health, decomposition of organic matter, crop yields, availability of the nutrients, etc., the microorganisms establish a range of ecological interactions that make the microbial populations develop or not (Vilatuña, 2019). These relationships are poorly studied and should be considered in research on soil microbiota (Goncharov & Tiunov, 2014; Jacoby et al., 2017; Erktan et al., 2020). When the microbial culture is performed, the growth of the entire community will not be observed because, perhaps, the growth of some species depends on others and once isolated in a different culture medium it does not grow; perhaps the new culture medium does not provide the necessary nutrients or perhaps the dilution factor used affected growth.
Traditional phenotypic identification schemes based on "observable" colony characteristics, such as morphology, development, and biochemical and metabolic properties, are essential as diagnostic methods for microbial community studies. However, these are insufficient since, from these methods, it is only possible to observe the growth of those facultative anaerobic microorganisms; but when it comes to environmental samples or samples taken from anaerobic systems, the use of other molecular techniques that contribute to quantification and microbial identification is recommended. It is important to determine exactly the relationship of the form and physiology of those microorganisms because it would help to understand their importance and function within the processes and how to manage them better.
Bacterial community structure and dynamics
In EM16 samples, 49 terminal fragments belonging to the Bacteria Domain were detected by TRFLP analysis.
Terminal fragments (TFs) with 62,181,404,476, 546, 571, 572, 575, 579, 582, 585, 597, 600, and 610 bp showed high relative abundance of more than 2% (Fig. 1). From the sugarcane straw soil isolated samples, 94 TFs were retrieved, where the fragments of 192,194,204,225,276,282,451,453, 472, 505, 508, 525, 546, 571, 575, 585, and 597 bp showed more than 2% relative abundance. From the bamboo soil isolated samples, 83 TFs were obtained, where the TFs of 152, 192, 194, 204, 225, 276, 281, 282, 451, 453, 472, 505, 508,525,546, and 579 bp showed a relative abundance more than 2% (Fig. 1). When the total communities are explored, including the TFs with a relative abundance below 2%, 13 TFs were found in both isolated soil samples (i.e., 192,194, 204,225,276, 282,451,453,472, 505, 508, 525, and 546 bp). That is, probably, because they are endemic microbiota from these niches (brown carbonate soils).
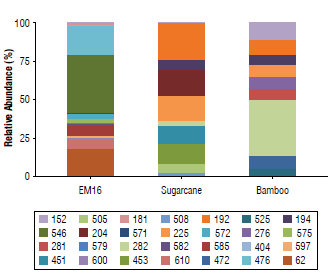
FIGURE 1 Relative abundance of bacteria terminal restriction fragments and their sizes (bp) in samples of EM16 and soil isolates (sugarcane crop and bamboo field). The relative abundance of each fragment was determined based on the height of its maximum of fluorescence in relation to the total of the heights of all the peaks detected in the sample.
According to Ion Torrent identification results from the Bacteria Domain (Tab. 1), five phyla were detected in the samples studied.
TABLE 1 Taxonomic distribution of the bacteria domain in samples of EM16 consortium and soil isolates identified by Ion Torrent technique.
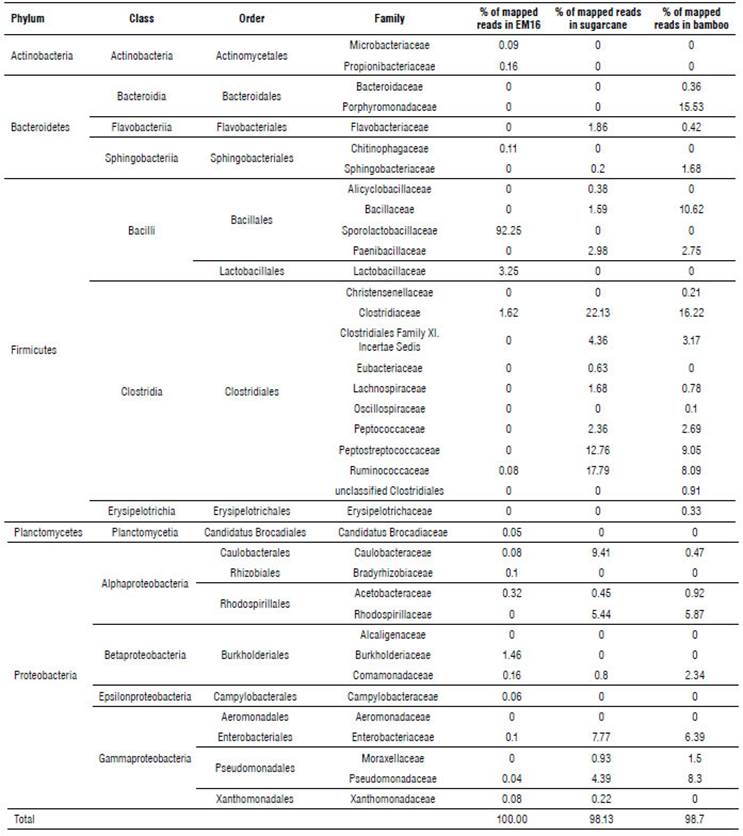
Almost 97% of the microbial community in EM16 was represented by the Firmicutes phylum. Other species were detected belonging to the phyla Proteobacteria (2.4%), Actinobacteria (0.25%), Bacteroidetes (0.11%), Planctomycetes (0.05%) (Tab. 1 and Fig. 2). A similar composition of microbial structure was identified in a thermoacidophilic EM consortium with six predominant phyla (Proteobacteria, Firmicutes, Chloroflexi, Bacteroidetes, Actinobac-teria, and Acidobacteria) (Henry et al., 2020). The result of this study showed that the microbial community in the EM16 consortium is favored by the formation of Sporolactobacillus species (family Sporolactobacillaceae 92.25%) (Fig. 2).
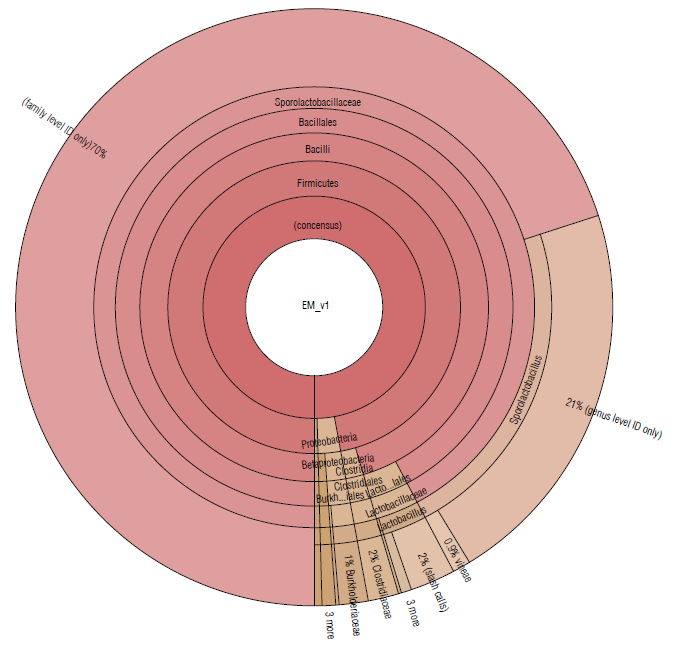
FIGURE 2 Krona plot of Identified bacteria in EM16 sample, through the Ion Torrent technique, by using the Ion Reporter® software. The Krona plot shows the taxonomic distribution from phyllum to species level and the associated abundance based on the percentage of mapped reads taking into acount the NGS repositories based on NCBI database.
The members of Sporolactobacillus genus are catalase-negative, microaerophilic, spore-forming, homofermen-tative, lactic acid-producing species and require mainly carbohydrate for growth (Chang et al., 2008). Although Sporolactobacillus species are typically isolated from soil, they are occasionally isolated from fermented or spoiled foods (Yanagida et al., 1987; Fujita et al., 2010). The presence of these species was expected in the EM16 consortium due to the composition of the substrate (made in part by whey) considering that Sporolactobacillus, as well as Lactobacillus, are common in all types of dairy products (Madigan et al., 2019). The presence of lactic acid bacteria related to the breakdown of cellulolytic and lignified organic materials has also been reported in EM (Salminen & von Wright, 2004; Joshi et al, 2019).
The families Lactobacillaceae (3.14%) and Clostridiaceae (1.62%) were less abundant in this consortium (Fig. 2). The presence of Clostridium species is essential for cellulose degradation (Madigan et al., 2019), therefore they could be used to improve the degradation of lignocellulosic residues. On the other hand, Lactobacillus spp. have potential effects for agricultural needs, so they can enhance soil properties since they show an antagonistic effect against different phytopathogenic agents in the soil, mainly due to the decrease in pH, production of peptides with antimicrobial activity such as class I bacteriocins, and nisin that is very active against gram-positive bacteria (Tanya & Leiva-Mora, 2019) . In addition, Lactobacillus spp. can enhance the trophic interactions (Quattrini et al., 2018; Naik et al., 2019), biotic and abiotic responses (Tsuda et al., 2016; Blainski et al., 2018; Abd El-Mageed et al., 2020; Muhialdin et al., 2020) , and plant growth and productivity (Daranas et al., 2018; Quattrini et al., 2018). The presence of these species is, therefore, essential in the EM16 consortium to enrich the soil microbiome and its functionality during the cycle of assimilation and de-assimilation of nutrients.
The taxonomic groups identified in EM16 are similar to other EM consortia (Alvarez et al., 2018; Naik et al., 2019); however, the diversity and richness of species depend on the environmental conditions and the substrate from which they come. The EM16 consortium could be an efficient alternative to use in sustainable agriculture since it can promote microbial consortia interaction in the soil and degrades organic components at the same time it supports a better micro or macronutrient assimilation by roots.
The sugarcane soil sample was represented by the Firmicutes (67.06%), Proteobacteria (29.64%), and Bacteroide-tes (3.38%) phyla (Tab. 1). The most abundant families were Clostridiaceae (22.13%), Ruminococcaceae (17.59%), and Peptostreptococcaceae (12.76%) (Fig. 3).
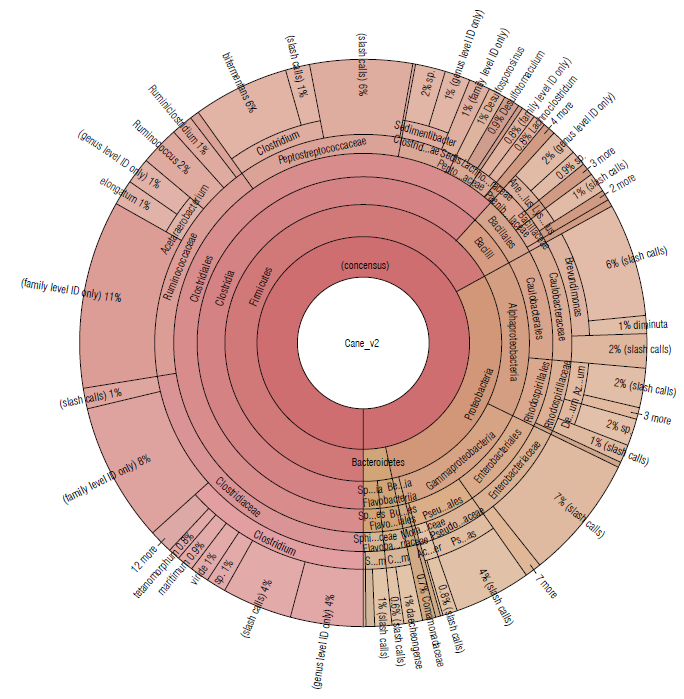
FIGURE 3 Krona plot of Identified bacteria in sugacane isolated soil sample, through the Ion Torrent technique, by using the Ion Reporter® software. The Krona plot shows the taxonomic distribution from phyllum to species level and the associated abundance based on the percentage of mapped reads taking into acount the NGS repositories based on the NCBI database.
A similar distribution of phyla was detected in bamboo soil samples, represented by Firmicutes (55.52%), Proteobacteria (26.28%), and Bacteroidetes (18.17%) (Tab. 1). The most abundant families were Clostridiaceae (16.22%), Porphyromonadaceae (15.53%), and Bacillaceae (10.62%) (Fig. 4).
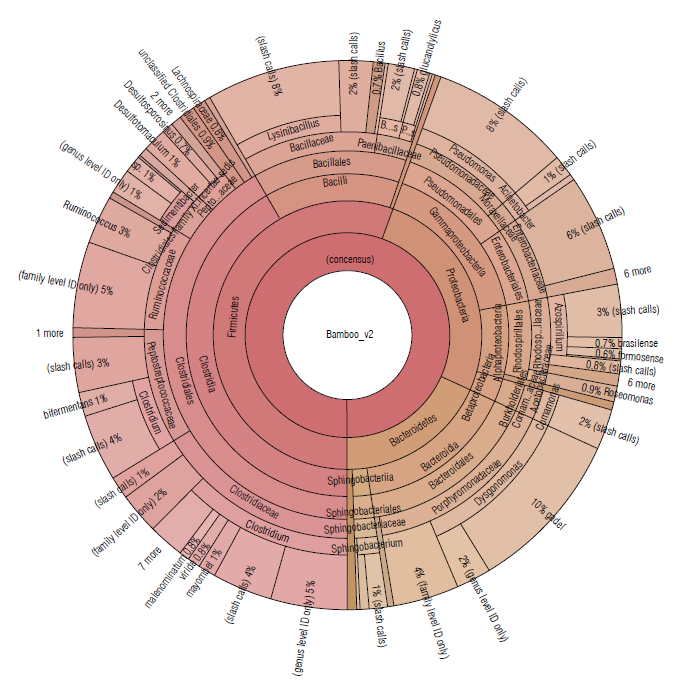
FIGURE 4 Krona plot of Identified bacteria in bamboo isolated soil samples using the Ion Torrent technique, using the Ion Reporter® software. The Krona plot shows the taxonomic distribution from the phyllum to species level and the associated abundance based on the percentage of mapped reads taking into acount the NGS repositories based on the NCBI database.
In the sugarcane and bamboo soil samples (Figs. 3-4), where bacterial species belonging to the phylum Firmicutes predominated, more than 10 different species of the Clostridium genus were identified to the species level. These are fermentative bacteria, strictly anaerobic with great metabolic diversity, gram-positive, endospore-forming, generally fermenting sugars or amino acids usually found in soil samples (Madigan et al, 2019). Besides, in the Clostridiales order, with an abundance greater than 1.5% the genera Ruminococcus and Sedimentibacter have been identified in both soil samples.
Ruminococcus species are defined as strictly anaerobic, gram-positive, non-motile cocci that do not produce endospores and require fermentable carbohydrates for growth (Goodfellow et al, 2009). They have been detected in greatest abundance in samples taken from herbivorous and omnivorous animal hosts, but a few have been found in environmental sources, generally in host-associated samples (La Reau et al, 2016). Some species are cellulolytic, playing an important role in the degradation of particulate substrates (Flint et al., 2008). Other members of family Ruminococcaceae are numerically abundant in the human intestinal tract, like bacterial species capable of degrading crystalline cellulose (Chassard et al., 2012; Morais et al., 2016). Others are non-cellulolytic and use polysaccharides like resistant starches (Ze et al., 2012) or selectively use various plant hemicelluloses (Wegmann et al., 2014).
Species of the genus Sedimentibacterhave been commonly isolated from anaerobic microbial communities (Breitenstein et al, 2002; Woo et al, 2004; Imachi et al, 2016). Cells are slightly curved rods, motile, gram-positive and spores might be formed. Growth is strictly anaerobic and requires yeast extract and is supported by the fermentation of pyruvate or amino acids in a Stickland-type reaction. Hydrogen is not produced and carbohydrates are not fermented (Breitenstein et al, 2002).
Members of the genus Pseudomonas (class Gammaproteobacteria) were also identified in soil samples from sugarcane and bamboo. These species show great metabolic diversity and consequently are widely distributed in nature. Some species are pathogenic for humans, animals, or plants. The metabolism of Pseudomonas is typically respiratory with oxygen as the terminal electron acceptor; but some species can also use nitrate as an alternative electron acceptor and carry out oxygen-repressible denitrification (dissimilatory reduction of nitrate to N2O or N2), allowing growth to occur anaerobically (Palleroni, 2015). They are typically oxidase and catalase-positive, do not form spores, with no gas formation from glucose, and perform catabolism of the carbohydrates by the alternative Entner-Doudoroff pathway and the cycle of tricarboxylic acids (Madigan et al, 2019).
The microbial diversity found in bamboo and sugarcane soil isolates could be used to enrich the EM16 consortium to achieve a more complete bioproduct (with a broader metabolic capacity).
The phylogenetic analysis coincides with the results of the TRFLP, where some fragments were detected in both soil samples. In general, species richness, diversity and the evenness index allows a comparison of isolated soil and EM16 consortium samples as shown in Table 2, considering the Ion Torrent and TRFLP analyses results. As expected, the richness, diversity, and Pielou index were higher in isolated soil samples compared to EM16 samples (Tab. 2) due to the dynamic conditions and the diversity of nutrients in these natural ecosystems. Despite the low diversity in the EM16 samples, it is worth noting the uniformity of the community with a coefficient of over 0.5.
TABLE 2 Richness, diversity, and evenness indexes calculated for each sample considering the Ion Torrent and TRFLP (in parenthesis) analysis results. Species with a relative abundance more than 1% were considered.
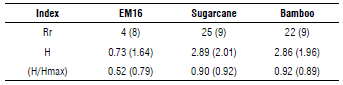
Rr=species richness, H=diversity index, H/Hmax=evenness index.
Despite the differences among the indices with respect to the richness, the tendency with respect to diversity and the evenness is similar for both techniques. That is why the authors consider that TRFLP is more useful for diversity and dynamic evaluation and Ion Torrent is used for deeper identification until species level.
Conclusions
This study provides a comprehensive morphological and molecular characterization of the efficient microorganisms (EM16) and sugarcane and bamboo soil samples. The diversity, dynamic and taxonomic identification achieved in this study for the EM16 showed the perspective for using these consortia for bioremediation, considering the wide metabolic pathway including the presence of key species like Lactobacillus sp. with high potentials for biodegradation of lignocellulosic resilient compounds. Future studies may be aimed at evaluating the mixture of these microorganisms to obtain a microbial consortium with a higher metabolic capacity and increased effectivity in its agricultural or environmental use compared to EM already studied. The use of microbiological and molecular tools under polyphasic approaches allows the completed characterization of non-cultivable microorganisms reported for the first time from an efficient microorganism consortium.

