










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkCiencia en Desarrollo
Print version ISSN 0121-7488
Ciencia en Desarrollo vol.7 no.1 Tunja Jan./June 2016
Hidrogenación electrocatalítica de oleato de metilo y linoleato de metilo con titanio recubierto con sistemas tipo Ni/SnO(x)
Electrocatalytic Hydrogenation of Methyl Oleate and Methyl Linoleate by Titanium covered with by /Ni/SnO(x) type Systems
A. S. Salinasa,*
J. V. Gutiérreza
F. E. Larmat G.b
A. E. Ramírez-Sanabriaa
a Grupo de Catálisis, Departamento de Química, Universidad del Cauca, Popayán, Colombia.
* Autor de correspondencia: assalinast@gmail.com.
b Grupo de Electroquímica, Universidad del Valle, Cali, Colombia.
Recepción: 03-dic-2015 Aceptación: 25-ene-2016
Resumen
Expone los resultados de la hidrogenación electrocatalítica (HEC) de los ésteres metílicos de los ácidos linoleico y oleico. El primer parámetro estudiado fue el potencial de electrólisis en el cual se genera el hidrógeno activo mediante voltametría cíclica, estableciéndose en -600 mV. Los electrocatalizadores empleados a base de titanio recubiertos con sistemas de níquel soportado sobre óxidos de estaño (II) y (IV) mostraron diferente comportamiento hacia los diversos substratos estudiados. El sistema con óxido de estaño (II) fue más activo hacia la hidrogenación de oleato de metilo, y el sistema con óxido de estaño (IV) resultó más activo frente a la hidrogenación de linoleato de metilo.
Palabras clave: Electrocatalizadores, hidrogenación electrocatalítica, metil ésteres, óxidos de estaño.
Abstract
This paper presents the Electrocatalytic Hydrogenation (ECH) of methyl oleate and methyl linoleate results electrolysis potential by which active hydrogen is producedby cyclic voltametry, was established at −600 mV. The electrocatalysts used were based on Titaniumcovered with nickel systems supported over tin oxide (II) and (IV) showed different behavior towards the diverse substrata studied. Tin oxide (II) system was most active towards the methyl oleate hydrogenation, whereas, tin oxide (IV) system was most active to methyl linoleate hydrogenation.
Key words: Electrocatalysts, Electrocatalytic Hydrogenation, Methyl esters, Tin Oxides.
1. Introducción
La hidrogenación de compuestos orgánicos es una reacción tipo RedOx cuyo resultado final es la adición de hidrógeno a un enlace múltiple; para que esta sea posible se deben cumplir tres requisitos: I) una fuente de hidrógeno, que en la mayoría de los casos corresponde a hidrógeno gaseoso; II) una temperatura, que puede oscilar entre 25 y 400 °C, y III) un catalizador (generalmente metales nobles o de transición soportados o no soportados o metales tipo Raney), sin el cual no habría reacción, pues este es el que permite la adición del hidrógeno al enlace insaturado [1].
En las últimas décadas, la hidrogenación electrocatalítica (HEC) ha surgido como un método alternativo para hidrogenar moléculas orgánicas insaturadas bajo condiciones más suaves (en comparación con las rutas químicas tradicionales), presión atmosférica y temperatura ambiente, ya que el hidrógeno activo es generado in situ por reacciones de descarga de protones en los sitios superficiales del electrocatalizador [2]. Para los aceites y sus derivados, se ha prestado una atención especial, dado el interés en obtener aceites libres de ácidos trans [3, 4].
Tradicionalmente, para la hidrogenación homogénea de los ésteres metílicos de ácidos grasos se han empleado como catalizadores metales nobles tipo Pt, Pd y Ru o complejos organometálicos de los mismos metales, los cuales son más activos a condiciones de temperatura y presión de hidrógeno (PH2) moderada. Sin embargo, debido a factores como su elevado costo y la dificultad de separar el catalizador al final de la reacción, se ha impulsado el desarrollo de catalizadores heterogéneos a partir de metales no preciosos como Cu, Fe o Ni (por ejemplo, el catalizador de Raney-Níquel, una aleación de Ni/Al) como alternativa; pero estos son activos solamente bajo condiciones de reacción más fuertes que pueden llevar a una baja selectividad, debido a que estas condiciones favorecen reacciones de isomerización del doble enlace Bailey (1949). En contraste, la hidrogenación electrocatalítica ha mostrado buenos rendimientos y selectividades con metales baratos como níquel, cobre o hierro, con las bondades de condiciones de reacción suaves [5-8].
Uno de los productos que se persigue obtener mediante la hidrogenación de ésteres son los alcoholes grasos, debido a sus aplicaciones en la industria de los tensoactivos (REF). No obstante, para poder reducir la función éster se hace necesario la incorporación de un segundo metal o promotor, por lo que últimamente se ha dirigido especial atención a metales como el estaño, por sus propiedades como ácido de Lewis y por su carácter anfótero, además de que se han obtenido resultados sobresalientes en obtención de alcoholes grasos insaturados [9]. En la figura 1 se indican los productos que puede generar la hidrogenación de oleato de metilo y linoleato de metilo, de los cuales se destacan la hidrogenación del enlace olefínico, que genera estearato de metilo; la reducción de la función éster, para dar el alcohol oleico; la reacción de isomerización del enlace doble, que lleva a la formación de elaidato de metilo, y la generación de linoleato de metilo por deshidrogenación.
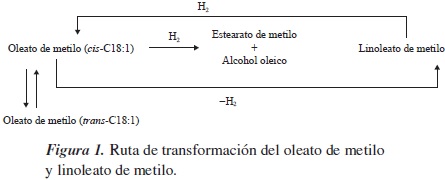
Dado el interés en el níquel como metal hidrogenante y en el estaño como promotor, en el presente trabajo se ha decidido estudiar la actividad de electrocatalizadores a base titanio recubiertos con sistemas de níquel soportados en óxidos de estaño (II) y (IV) para la hidrogenación por vía electrocatalítica de los ésteres grasos, oleato de metilo (C18:1) y linoleato de metilo (C18:2).
2. Sección experimental
2.1. Obtención de los electrocatalizadores
Obtención de los soportes. Se empleó el método de precipitación controlada para obtener los óxidos de estaño (II) y (IV) [10]. Para ello, el precursor, cloruro de estaño (II) (100%, Mallinckrodt), en concentración 0,1 M en ácido acético glacial (99,7%, EM) y etanol absoluto (99,9%, Merck), respectivamente, se trata con hidróxido de amonio concentrado (28%, J.T. Baker) suministrado mediante un dosificador automático Dosimat (Metrohm), hasta alcanzar pH 8 y 6,25, a los cuales se asegura la obtención de los óxidos de estaño (II) y (IV), respectivamente. Los soportes se lavaron con etilendiamina 2 M y se secaron a70 °C.
Impregnación de níquel. Una vez obtenidos, los soportes de óxido de estaño (II) y (IV) se impregnaron por vía húmeda, adicionando una disolución de cloruro de níquel (98%, Merck) 0,1 M bajo agitación magnética (200 rpm), hasta obtener sólidos con contenido aproximado de níquel 2-3% en peso. Se evaporó a 60 °C en plancha de calentamiento (Corning) con agitación y luego a 105 °C en un horno (WTC Binder) por 24 horas.
Recubrimiento de los electrodos. Los electrocatalizadores se obtuvieron mediante la técnica screen printing; para ello se formó una pasta viscosa, mezclando glicerina (99,9%, Fischer) con los sólidos a base de níquel. Esta mezcla se esparció uniformemente con ayuda de un pincel sobre placas de titanio de 1 cm x 1 cm (99,6%, Goodfellow). Las placas recubiertas se secaron a 60 °C durante 24 horas, y, luego, fueron calcinadas a 390 °C (SnO) y 450 °C (SnO2) durante 8 horas en atmósfera estática de aire. Posteriormente, el sistema fue reducido en flujo continuo de hidrógeno a 400 °C en una autoclave (Parr Instrument Company) durante media hora.
2.2. Caracterización de los sistemas Ni/SnO(x)
Para cuantificar el contenido de níquel se utilizó la espectrofotometría de absorción atómica de llama; esto se hizo en un equipo Thermo Solaris a una longitud de onda de 232 nm, previa disolución de los catalizadores en H2SO4 (Merck, 96%). El área superficial se calculó empleando el método BET mediante adsorción de nitrógeno a −77 °C y posterior desorción, en un equipo Quantachrome Chembet 3000. También se realizó difracción de rayos X en polvo, para lo cual se empleó un difractómetro de rayos X, Rigaku Miniflex II con geometría Bragg Brentano, equipado con una fuente de radiación de Cu λ Kα de 0.1540562 nm de longitud de onda que opera a 30 kV y 15 mA con un detector de NaI que no emplea monocromador. Los patrones de difracción (PXRD) fueron recolectados en el intervalo de 10 a 90° en 2 Theta a una velocidad de barrido de 2°/min a condiciones de ambiente.
2.3. Determinación del potencial de electrolisis
El potencial de electrólisis, al cual se genera el hidrógeno activo, se determinó mediante voltamperometría cíclica, con una solución de ácido sulfúrico (96%, Merck) 0,1 M como electrolito soporte en agua deionizada: etanol absoluto en relación 1:40, empleando un potenciostato/galvanostato (Electrochemical Instruments) y una celda electroquímica con tres electrodos: como electrodo de trabajo se empleó el electrocatalizador fabricado anteriormente, el electrodo de referencia fue de Ag/AgCl y como electrodo auxiliar se empleó una lámina de platino (99,95%, Goodfellow) de 1 cm x 1 cm. Se realizaron 20 ciclos entre −800 y 400 mV y, luego, desde 400 a −800 mV, a una velocidad de barrido de 50 mV/s.
2.4. Hidrogenación electrocatalítica
La hidrogenación electrocatalítica fue llevada a cabo a temperatura y presión atmosférica en una celda no dividida con1gde oleato de metilo (80% C18:1, C18:2, Sterenearie Dubois) disuelto en 50 mL de una solución de ácido sulfúrico 0,1M. El sistema fue purgado con nitrógeno (99,999% Cryogas) durante 3 minutos, se tomó una alícuota como punto cero de la reacción e, inmediatamente, se llevó a cabo la electrólisis por 6 horas a −600 mV.
2.5. Análisis por cromatografía de gases
La reacción fue seguida cada 2 horas; para ello se tomó una alícuota que fue pasada a través de filtros de 0,45 μm (Millipore). Se inyectó un volumen de 2,0 μL en un cromatógrafo de gases Shimadzu C14-A provisto de detector de ionización de flama, unidad integradora Chromatopac C-R8A (Shimadzu) y una columna capilar SGE ID-BP20 de 25 m × 0,32 mm × 0,25 μm. Para la columna se empleó el siguiente programa de temperatura, 160 °C (1 min), 30 °C/min, 190 °C (1 min), 10 °C/min, 210 °C(1 min), 10 °C/min, 240 °C (2 min); la temperatura del inyector y del detector fue de 260 °C y 280 °C, respectivamente.
3. Resultados y Discusión
3.1. Caracterización de los sistemas Ni/SnO(x)
Una vez se obtuvieron los sistemas a base de níquel soportado en los óxidos de estaño (II) y (IV), fueron caracterizados por varias técnicas. La absorción atómica de llama se empleó para determinar el contenido de níquel presente en los sólidos; los resultados para los sistemas obtenidos se detallan en la tabla 1.

Se aprecia que las cantidades que se requieren dopar de níquel están entre 2,0 y 3,5% respecto al soporte. El primer resultado obtenido de los análisis de área superficial, tabla 1, es que los dos soportes presentan una diferencia considerable en el valor de sus áreas superficiales específicas, pues la del SnO2 es prácticamente tres veces la del SnO. El valor del área para el soporte muestra una reducción luego de impregnar con Níquel, lo cual se explica por el tratamiento térmico posterior a la impregnación de la fase metálica.
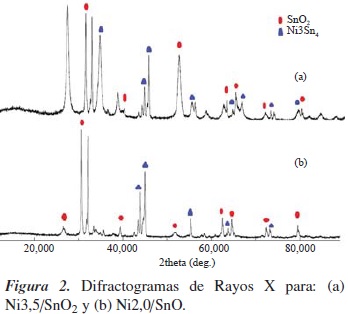
En la figura 2(a), correspondiente al sistema Ni3,5/SnO2, se aprecia que la estructura cristalina del soporte con estructura tipo rutilo permanece invariable, pero se obtuvieron nuevas fases cristalinas correspondientes a especies tipo cluster formado por el níquel y el estaño de formula Ni3Sn4, de acuerdo con la carta cristalográfica 4-851, promovidos por fenómenos de difusión de átomos de estaño del soporte. En el caso del catalizador Ni2,0/SnO, figura 2(b), su difractograma muestra que al soportar níquel no se altera la estructura cristalina del soporte, al menos de manera significativa, pues se aprecia que los desplazamientos 2θ, característicos de la fase cristalina del óxido, permanecen constantes, aunque aparecen nuevas especies relacionadas con una fuerte interacción metal-soporte, tipo cluster de Ni3Sn4, tal como se presentó en el caso de Ni3,5/SnO2. La formación de dichas especies puede atribuirse a una mayor dispersión de níquel, y pueden brindar ciertas propiedades al sistema catalítico.
3.2. Determinación del potencial de electrólisis
La especie activa en la reacción de hidrogenación electrocatalítica, el hidrógeno atómico, debe ser generado in situ a bajos sobrepotenciales por la electrorreducción del ión hidronio, el cual reacciona con el substrato orgánico insaturado [11]. Manteniendo fijas la composición de la disolución y la temperatura del sistema, se estudió que no hubiese algún de tipo de inflencia del solvente, agua o etanol, en la generación de hidrógeno atómico, pues estos son de naturaleza prótica. El experimento se llevó a cabo, en primer lugar, en ausencia de electrolito soporte, H2SO4, en los dos electrocatalizadores: a base de SnO2 y a base de SnO. La figura 3(a) muestra el voltamograma cíclico obtenido de este ensayo; en él no se aprecian picos significativos relacionados con algún proceso RedOx del solvente. Al realizar el ensayo en presencia de H2SO4 con el sistema Ni3,5/SnO2 se obtiene el voltamograma cíclico de la figura 3(b), tras 20 ciclos entre −800 y 400 mV, en él se aprecian dos picos, uno a −350 mV, en el barrido anódico  , y otro a −600 mV, en el barrido catódico
, y otro a −600 mV, en el barrido catódico  . El pico generado en el barrido catódico en un valor de potencial de −600 mV se relaciona con la reacción de descarga del protón presente en la disolución y mediante la cual se genera la especie activa de hidrógeno (H+ → H.), luego del cual se nota una fuerte caída de la corriente por la formación de hidrógeno molecular en el cátodo. El pico presente en el barrido anódico del voltamograma se asocia con la reacción inversa, la oxidación del hidrógeno atómico.
. El pico generado en el barrido catódico en un valor de potencial de −600 mV se relaciona con la reacción de descarga del protón presente en la disolución y mediante la cual se genera la especie activa de hidrógeno (H+ → H.), luego del cual se nota una fuerte caída de la corriente por la formación de hidrógeno molecular en el cátodo. El pico presente en el barrido anódico del voltamograma se asocia con la reacción inversa, la oxidación del hidrógeno atómico.
Al pasar al análisis del voltamograma obtenido con el sistema Ni2,0/SnO, figura 3(c), se nota la presencia de los mismos picos obtenidos con el sistema Ni3,5/SnO2, de lo que se puede inferir que no hay una influencia significativa de la naturaleza del óxido de estaño sobre el potencial de la reacción de descarga del protón, más bien que esta se da en la superficie de las partículas metálicas de níquel.

Por estar implicado en el barrido catódico, el pico a −600 mV se toma como el sobrepotencial de reducción del hidrógeno, en el cual se genera la especie activa que es adicionada a la insaturación del substrato orgánico. El potencial obtenido resulta bajo en comparación con otros reportados con electrodos de níquel, como en el trabajo de da Silva et al. [12], quienes alcanzaron un potencial de −1,6 V. Se debe ser cuidadoso con la selección de este potencial, pues podría presentarse la evolución de hidrógeno gaseoso que limita la eficiencia de la hidrogenación [13].
3.3. Reacción de hidrogenación electroquímica con el sistema Ni/SnO
Dado que el rendimiento de la reacción es dependiente del material catódico, se evaluaron dos sistemas, Ni/SnO y Ni/SnO2, sobre electrodos de titanio en la hidrogenación de oleato de metilo y linoleato de metilo. La figura 4 muestra el esquema reaccional para los dos substratos.

En la figura 5(a) se aprecian los perfiles de conversión de linoleato de metilo y oleato de metilo con el electrocatalizador Ni2,0/SnO; se puede notar una mayor actividad hacia la hidrogenación del oleato de metilo desde al inicio de la reacción, llegándose a una conversión del 82%. En contraste, el linoleato de metilo solo comienza a ser convertido a la mitad del tiempo de reacción, y lo máximo que se consigue es un 8% de conversión. Esto muestra una selectividad de este catalizador, influenciada por las características del soporte.

3.4. Reacción de hidrogenación electroquímica con el sistema Ni/SnO2
El electrocatalizador Ni3,5/SnO2 mostró un comportamiento totalmente diferente al anterior; tal como se aprecia en la figura 5(b), predominó la hidrogenación de linoleato de metilo, pero solo desde la mitad del tiempo de reacción, con una leve actividad al comienzo, en tanto el oleato de metilo no fue convertido; esto habla de una mayor afinidad de este sistema hacia el substrato linoleato, que al tener dos insaturaciones, en posiciones 9 y 12 en la cadena carbonada, debe presentar una interacción diferente en comparación al oleato por los sitios de adsorción del sólido.
3.5. Estabilidad de los recubrimientos
Con el fin de evaluar la estabilidad estructural del recubrimiento del electrocatalizador, se realizó un análisis por difracción de rayos X del sistema Ni3,5/SnO2, antes y luego de 6 horas de reacción con oleato de metilo. Como el procedimiento es llevado a cabo en medio ácido, este puede provocar alguna corrosión del material catódico. La figura 6 muestra los difractogramas tomados a los electrocatalizadores antes y después de reacción, 6(a) y 6(b), respectivamente. Se puede observar que no se presenta modificación alguna de tipo estructural en el recubrimiento tras su aplicación en la reacción, pues las fases cristalinas más importantes asociadas a la estructura tetragonal tipo rutilo del SnO2 permanecen constantes; igualmente, no se aprecia algún patrón de difracción asociado al titanio, indicando la alta estabilidad del recubrimiento. Particularmente, esto puede abrir la posibilidad de que los sistemas electrocatalíticos puedan ser reutilizados en varias reacciones.

4. Conclusiones
La metodología de hidrogenación electrocatalítica permite el uso de materiales relativamente económicos, como titanio, níquel y óxidos de estaño, para su aplicación con éxito en la obtención de compuestos de interés industrial. Particularmente, se hace importante por el uso de condiciones de reacción suaves, lo que favorece un ahorro en costos operacionales y energéticos, siendo un método ambientalmente atractivo.
En la hidrogenación electrocatalítica de oleato de metilo y linoleato de metilo, con sistemas electrocatalíticos de titanio recubierto de níquel soportado sobre óxidos de estaño (II) y (IV), se observó un comportamiento totalmente diferente para estos dos substratos; con el soporte SnO prevaleció la hidrogenación de oleato de metilo, mientras que cuando se usó SnO2 como soporte se favoreció la hidrogenación de linoleato de metilo.
Agradecimientos
Al Laboratorio de Materiales Nanoestructurados y Funcionales de la Universidad Nacional de Colombia, sede Manizales, a cargo del Dr. Oscar H. Giraldo, por su cooperación en los difractogramas de rayos X. A la Universidad del Cauca, por el soporte financiero dado al proyecto. A la Universidad del Valle, por el empleo del portenciostato-galvanostato.
Referencias
[1] C. M. Cirtiu, A. B. Wittmeyer, and H. Menard, "Comparative study of catalytic and electrocatalytic hydrogenation of benzophenone", Catalysis Communications, vol. 8, pp. 751-754, 2007. [ Links ]
[2] R. C. Z. Lofrano, J. M. Madurro, L. M. Abrantes, and J. R. Romero, "Electrocatalytic hydrogenation of carbonilic compounds using an electrode with platinum particles dispersed in films of poly-[allyl ether p-(2-aminoethyl) phenol] co-polimerized with allyl phenyl ether", Journal of Molecular Catalysis A: Chemical. vol. 218, pp. 73-79, 2004. [ Links ]
[3] Mondal & Lalvani (2003a) [ Links ]
[4] Mondal & Lalvani (2003b) [ Links ]
[5] J. Lessard, "Reduction of alkenes and conjugated alkenes", J. R. Sowa. Catalysis of Organic Reaction, Ed., Taylor y Francis Group, Boca Raton, pp. 3-18, 2005. [ Links ]
[6] Pintauro et al. [ Links ]
[7] An et al. (1998) [ Links ]
[8] Yusem et al. (1996) [ Links ]
[9] Y. Pouilloux, A. Piccirilli, and J. Barrault, "Selective hydrogenation into oleyl alcohol of methyl oleate in the presence of Ru-Sn/Al2O3 catalysts", Journal of Molecular Catalysis A: Chemical, vol. 108, pp.161-166, 1996. [ Links ]
[10] C. Ararat. "Sintesis de SnO2 por (MPC) y su uso como catalizador o soporte catalitico de Sn, Ni o Sn-Ni en reacciones de transesterificación del aceite de palma", Trabajo de grado, Universidad del Cauca, 2005. [ Links ]
[11] H. Ilikti, N. Rekik, and M. Thomalla. "Electrocatalytic hydrogenation of alkyl-substituted phenols in aqueous solutions at a Raney nickel electrode in the presence of a nonmicelleforming cationic surfactant". Journal of Applied Electrochemistry. vol. 34, pp. 127136, 2004. [ Links ]
[12] J. J. G. da Silva, M. O. F. Goulart, and M. Navarro, "Electrocatalytic hidrogenation of diethyl fumarate. A simple system development", Tetrahedron, vol. 55, pp. 7405-7410, 1999. [ Links ]
[13] D. S. Santana, M. V. F. Lima, J. R. R. Daniel, and M. Navarro, "Electrocatalytic hydrogenation of organic compounds using current density gradient and sacrificial anode of nickel", Tetrahedron Letters, vol. 44, pp. 4725-4727, 2003. [ Links ]

