











 text in
text in  Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCCIÓN
El caracol rosado, Lobatus gigas, es una especie de importancia ecológica y económica en toda su área de distribución (Catarci, 2004). Esta especie juega un papel importante en las interacciones ecológicas de las poblaciones marinas cercanas a la costa, como uno de los principales herbívoros de algas marinas, algas epífitas y detritus. Además, es un organismo comensal, sirve como fuente de alimento para una amplia variedad de organismos marinos y compite con ellos por los recursos (Stoner and Waite, 1991; Tewfik, 1997; Catarci, 2004). Asimismo, es un recurso pesquero con valor económico en el comercio de alimentos marinos. Por lo tanto, muchos países deben establecer políticas para regular su explotación comercializándolo de manera sostenible sin poner en peligro su conservación. No obstante, a pesar del diseño y ejecución de medidas de gestión nacionales e internacionales para controlar la sobreexplotación y el acceso a este recurso, (Prada et al., 2008), su densidad poblacional continúa disminuyendo (Ballesteros et al., 2005; Stoner et al., 2012). Esta situación dificulta el mantenimiento adecuado de la población, lo que afecta a su capacidad de recuperación y hace que su comercialización sea insostenible (Stoner and Ray-Culp, 2000). En consecuencia, se ha incluido en el apéndice II de la Convención sobre el Comercio Internacional de Especies Amenazadas de Fauna y Flora Silvestres (CITES) (Daves and Fields, 2004). Se han establecido varias alternativas para superar esta situación y recuperar las poblaciones sobreexplotadas de L. gigas, como la creación de áreas marinas protegidas (AMP) (Appeldoorn, 1994; Stoner, 1996; Anon, 1999) que permitan preservar la alta densidad del stock de desove y el mantenimiento de un refugio para los adultos con mayor capacidad de reproducción (Anon, 1999). Otra alternativa es la acuicultura con técnicas dirigidas a la producción de juveniles (Brownell, 1977; Creswell, 1994).
En la Reserva de la Biosfera de Seaflower, se han establecido varias medidas de conservación para regular la pesca de L. gigas y mejorar la comprensión de su biología (Castro et al., 2007). Más específicamente, los estudios se han centrado principalmente en la genética poblacional (Landínez- García et al., 2011; Márquez et al., 2012), la dinámica y la biología reproductiva de este molusco (Aranda et al., 2001; Aranda et al., 2003a, b; Delgado et al., 2004; Castro et al., 2007). Sin embargo, estas alternativas de conservación no han tenido en cuenta los estudios sobre la microbiota, que es un aspecto fundamental del éxito del cultivo y la recuperación de organismos marinos. Por ejemplo, se ha demostrado en varias especies de peces y crustáceos que la comunidad microbiana influye en diversas funciones del hospedador, incluyendo el desarrollo, la digestión, la nutrición, y la resistencia e inmunidad a las enfermedades (Merrifield and Ringø, 2014), aspectos fundamentales para la salud de cualquier organismo. A pesar de esto, hasta la fecha se sabe muy poco acerca de cómo la microbiota influye en el crecimiento y desarrollo del caracol rosado (Kirjavainen and Gibson, 1999; Kowalik et al., 2006, Pérez et al., 2014). Recientemente, otros autores han analizado la microbiota del caracol rosado por medio de métodos dependientes de cultivo, identificación genotípica (Acosta et al., 2009, Pérez et al., 2014) y otros métodos independientes de cultivo (Pérez et al., 2014) utilizando muestras de intestino y de la secreción del caracol silvestre recolectadas cerca de la Isla del Rosario e Isla Tortugas en Colombia. Estos estudios demostraron la presencia de Pseudoalteromonas sp., Halomonas sp., Psychrobacter sp., Cobetia sp., Pseudomonas sp., Vibrio sp. y Burkholderia sp. (Acosta et al., 2009; Pérez et al., 2014). Cuartas et al., (2018), utilizando el análisis histológico, pirosecuenciación 454, y una combinación de amplificación por reacción en cadena de la polimerasa (PCR por sus siglas en inglés) y secuenciación Sanger indicaron que el agente etiológico de las protrusiones musculares es un parásito perteneciente a la subclase Digenea y está asociado a clados de bacterias y de hongos. Sin embargo, actualmente, no existen informes sobre la caracterización molecular de la microbiota bacteriana asociada a los compartimentos de la gónada y el tracto digestivo de L. gigas.
El tracto digestivo es un ecosistema mixto que contiene un consorcio de microorganismos dinámico y complejo, que desempeña un papel clave en la nutrición y salud del hospedador (Bäckhed et al., 2004). La microbiota intestinal está involucrada en procesos importantes, como la estimulación de la proliferación epitelial, el desarrollo del metabolismo de nutrientes y la respuesta inmune innata (Rawls et al., 2004; Sommer y Bäckhed, 2013; Thaiss et al., 2016). Por lo tanto, el conocimiento sobre las bacterias asociadas con los diferentes compartimentos del aparato digestivo podría ser útil para controlar la microbiota como estrategia para mejorar la nutrición, prevenir las infecciones por patógenos y desarrollar nuevas metodologías que contribuyan al éxito del cultivo de esta especie. Hasta la fecha, no hay estudios dirigidos a la identificación de las comunidades bacterianas que habitan la gónada del caracol y el papel potencial que desempeñan en la salud y el desarrollo de esta especie. No obstante, se ha reportado la presencia de bacterias en la gónada de otros moluscos, como Argopecten purpuratus donde se encontró la presencia permanente de los géneros Vibrio, Acinetobacter, Moraxella, Pseudomonas y Cytophaga en los órganos reproductivos (Chavez y Riquelme, 1994; Riquelme et al., 1995a). En el molusco Dendrodoris nigra, se determinó la presencia de bacterias en la gónada mediante microscopía electrónica, pero las mismas no fueron clasificadas (Klussmann-Kolb y Brodie, 1999). Aunque la microbiota gonadal es poco conocida en los organismos marinos, estudios previos del microbioma en mamíferos han demostrado que existe simbiosis entre bacterias y mamíferos. Estas bacterias desempeñan papeles importantes, como proporcionar protección contra la colonización de patógenos y producir compuestos antimicrobianos (Reid et al., 2011; Miller et al., 2017; Kindinger et al., 2017).
El objetivo principal del presente trabajo fue determinar la estructura de la comunidad bacteriana asociada a la gónada y a las diferentes secciones del intestino de L. gigas silvestres de la Reserva de la Biosfera de Seaflower del Caribe, utilizando un método dependiente de cultivo y otro independiente de cultivo. Además, analizamos la microbiota de muestras combinadas e individuales para estudiar la variación interindividual. Hasta donde sabemos, el presente trabajo es el primer examen de la microbiota bacteriana de la gónada y de todos los compartimentos intestinales del caracol rosado. Este enfoque permite la detección de las comunidades bacterianas dominantes que podrían tener relevancia en la reproducción y el desarrollo del caracol rosado.
MATERIALES Y MÉTODOS
Recolección y procesamiento de muestras
Veintidós muestras de L. gigas (caracol rosado), especímenes juveniles de aproximadamente 350 g cada uno, fueron recolectadas por pescadores en el Caribe colombiano, en el archipiélago de San Andrés (12°-16°N, 78°-82°O). Las muestras fueron proporcionadas por la Gobernación del Archipiélago de San Andrés, Providencia y Santa Catalina, a través del convenio de cooperación científica # 083/2012 (Cuartas et al., 2018). Los caracoles fueron transportados al laboratorio en hielo seco, donde se extrajo la gónada (G), el intestino delgado (ID) e intestino grueso (IG) de cada caracol de forma aséptica en frío. Los tejidos extraídos de 15 caracoles con un peso similar se separaron en cinco grupos y cada grupo se homogeneizó y se denominó pool (P) de cada tejido. La combinación de muestras es una práctica común para estudiar la microbiota intestinal en los peces (Hovda et al., 2007; Andlid et al., 1998; Romero and Navarrete, 2006). Los tejidos correspondientes a las siete (7) caracoles restantes se procesaron individualmente (I), para estudiar la variación interindividual (Romero y Navarrete, 2006). Finalmente, las muestras obtenidas de los tejidos (P e I) se maceraron inmediatamente con nitrógeno líquido y luego se almacenaron a -80ºC hasta su procesamiento. Debido a que las comunidades bacterianas se obtuvieron de cada compartimento intestinal que contenía epitelio y alimentos digeridos, la microbiota analizada fue una combinación de bacterias autóctonas (capaces de colonizar la superficie epitelial o moco del intestino del hospedador) y alóctonas (transitorias o asociadas a la digestión) (Figura 1).
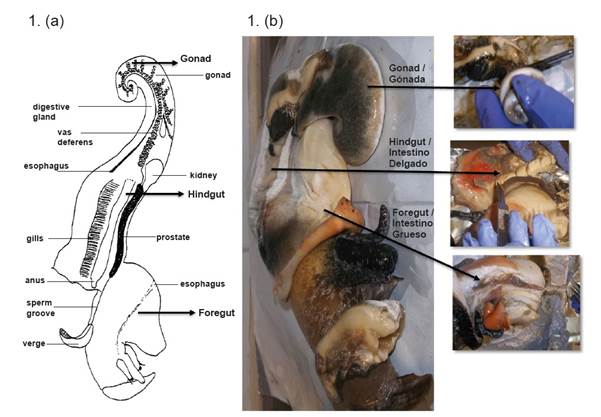
Figura 1 (a). Esquema de la anatomía de Lobatus gigas tomado de Reed (1996). (b). Disección y procesamiento de la gónada (G), intestino delgado (ID) e intestino grueso (IG) de L. gigas.
Para el método dependiente de cultivo, se sembraron diluciones en serie de homogeneizados de caracoles P o I en placas de agar marino (Difco) y agar tiosulfato-citrato-sales biliares-sacarosa (TCBS, Merck), y luego se incubaron las placas aeróbicamente a 20ºC por 2 días. Las unidades formadoras de colonias (UFC) recuperadas de los diferentes medios se conservaron en un 20% de glicerol a -80ºC y se mencionaron como fracción cultivable (C). La fracción directa (D) se refiere a las muestras de tejido macerado congeladas a-80ºC sin ser cultivadas (G, ID e IG). La nomenclatura de las muestras se resume en la Tabla 1.
Tabla 1 Nomenclatura de las muestras obtenidas de los compartimentos del intestino y la gónada de Lobatus gigas de la Reserva de la Biosfera de Seaflower. D=ADN total de la fracción directa, C=ADN de la fracción cultivable, I=muestras individuales, P=pools, G=Gónada, ID=intestino delgado e IG=intestino grueso.
| Nomenclatura Nomenclature | Extracción de ADN DNA Extraction | Orígenes Origen | Proceso Process | Muestras Samples |
|---|---|---|---|---|
| CIDI / CFI | Fracción cultivable "C" / Cultivable fraction “C” | Intestino delgado "ID" Foregut “F” | Individual “I” | CIDI2, CIDI3, CIDI4, CIDI6, CIDI16 CFI2, CFI3, CFI4, CFI6, CFI16 |
| CIDP / CFP | Pool "P" | CIDP1, CIDP2, CIDP4, CIDP5 CFP1, CFP2, CFP4, CFP5 | ||
| CIGI / CHI | Intestino grueso "IG" Hindgut “H” | Individual “I” | CIGI2, CIGI3, CIGI6, CIGI16 CHI2, CHI3, CHI6, CHI16 | |
| CIGP / CHP | Pool "P" | CIGP1, CIGP2, CIGP4, CIGP5 CHP1, CHP2, CHP4, CHP5 | ||
| CGI / CGI | Gónada "G" Gonad “G” | Individual “I” | CGI3, CGI4, CGI6, CGI16 | |
| CGP / CGP | Pool "P" | CGP1, CGP2, CGP4, CGP5 | ||
| DIDI / DFI | Fracción directa "D" / Direct fraction “D” | Intestino delgado "ID" Foregut “F” | Individual “I” | DIDI2, DIDI3, DIDI6, DIDI16 DFI2, DFI3, DFI6, DFI16 |
| DIDP / DFP | Pool "P" | DIDP1, DIDP2, DIDP3, DIDP4, DIDP5 DFP1, DFP2, DFP3, DFP4, DFP5 | ||
| DIGI / DHI | Intestino grueso "IG" Hindgut “H” | Individual “I” | DIGI2, DIGI3, DIGI6, DIGI16 DHI2, DHI3, DHI6, DHI16 | |
| DIGP / DHP | Pool "P" | DIGP1, DIGP2, DIGP3, DIGP4, DIGP5 DHP1, DHP2, DHP3, DHP4, DHP5 | ||
| DGI / DGI | Gónada "G" Gonad “G” | Individual “I” | DGI3, DGI4, DGI6, DGI16 | |
| DGP / DGP | Pool "P" | DGP1, DGP2, DGP3, DGP4, DGP5 |
Extracción de ADN
El ADN de la fracción cultivable (C) de cada muestra se extrajo con un kit de purificación de ADN a partir de sangre y tejido DNEasy (Qiagen, Düsseldorf, Alemania) utilizando un protocolo de extracción de ADN de DNEasy modificado para asegurar la lisis de las bacterias Gram positivas y Gram negativas, según lo descrito por Li et al. (2009).
La extracción del ADN genómico total de la fracción directa (D) de cada tejido se realizó utilizando un kit de extracción de ADN en suelo Ultraclean previa lisis tisular, como describen Ó Cuív et al. (2011). Brevemente, cada tejido diseccionado de L. gigas se obtuvo como se ha descrito anteriormente, se homogeneizaron 300 mg con 800 μl de tampón de lisis (6 mM Tris-HCl [pH 8], 100 mM EDTA, 1M NaCl) y se incubaron a 75ºC durante 10 minutos para inactivar las nucleasas. Se añadieron 20 μl de lisozima (200 mg/ml) y se incubaron las muestras durante toda la noche a 37ºC. Posteriormente, se realizó una digestión con 20 μl de proteinasa K (20 mg/ml) a 56ºC hasta la lisis completa de cada tejido.
Amplificación por PCR
Para obtener la huella molecular de la comunidad bacteriana del intestino delgado, el intestino grueso y la gónada mediante cultivo en agar marino (C), el gen RNAr 16S entre la región V3 y V6 se amplificó con iniciadores específicos 341F con una terminación extra de GC (5’ GCCTACGGGAGGCAGCAG 3’), y 907R (5' CCGTCAATTCMTTTGAGTTT 3') (McCracken et al., 2001). La PCR se llevó a cabo siguiendo las condiciones descritas por García et al. (2016).
La amplificación de la región hipervariable entre las regiones V3 y V6 del gen RNAr 16S de la fracción D se realizó con una PCR anidada. La primera PCR se llevó a cabo para amplificar la secuencias del gen RNAr 16S de longitud casi completa con iniciadores universales 27F (5`AGAGTTTGATCMTGGCTCAG 3`) y 1492R (5` GTTACCTTGTTACGACTT 3`) siguiendo las condiciones descritas por Espejo and Romero (1997). Luego, se usaron 2,5 μl del producto de la primera PCR como molde de ADN en la segunda reacción, que se realizó con los iniciadores específicos 341F(GC) y 907R (como se ha descrito previamente para las muestras cultivables) y siguiendo las condiciones reportadas por Espejo et al. (1998).
Análisis de electroforesis en gel de gradiente temporal de temperatura (TTGE, por sus siglas en inglés)
Se determinó la estructura de las comunidades bacterianas asociadas con el intestino delgado, el intestino grueso y la gónada mediante la técnica TTGE, que permitió separar fragmentos del gen RNAr 16S de aproximadamente 585 pb, con la suposición de que cada banda observada en los perfiles representa una especie bacteriana.
Los productos de la PCR obtenidos de ambas fracciones, usando los iniciadores 341F(GC) y 907R, se separaron mediante TTGE usando el Sistema Universal de Detección de Mutaciones DCode (BioRad, EE. UU.) en poliacrilamida al 7% (p/v), geles de urea 7 M en tampón TAE 1,25X durante 15 horas a 55V. La temperatura inicial y final fue de 66 ºC y 69 ºC, respectivamente, con un incremento de temperatura de 0,1ºC por hora. Los geles se tiñeron con AgNO3 (AMRESCO, OH, USA) (Sanguinetti et al., 1994). Los perfiles obtenidos para cada muestra estudiada se analizaron usando el programa GelCompar II (Applied Maths TX, USA) (Rademaker y De Bruijn, 2008, Garcia et al., 2016). Los carriles de referencia de cada muestra se alinearon en base a una escala de 100 pb, y se realizó un análisis de agrupamiento usando el coeficiente DICE (Nei and Li, 1979) y el método de agrupación de pares no ponderados con medias aritméticas (UPGMA, por sus siglas en inglés) (Mohammadi y Prasanna, 2003).
Análisis de la comunidad bacteriana
Se utilizó el programa GelCompar II (Applied Biosystems, Bélgica) para analizar los perfiles de TTGE a partir de las muestras de ADN. Para este análisis, se utilizó la matriz de presencia y ausencia para generar un análisis de similitud (ANOSIM, por sus siglas en inglés) basado en el índice de Bray-Curtis, utilizando el programa PAST versión 2.0 (Hammer et al., 2001). Se evaluaron las diferencias significativas de la diversidad bacteriana con la opción "Diversity t-test" del programa PAST. Para este análisis, se consideró que el número de banda representaba el número de especie o Taxón (S) y la intensidad de la banda la abundancia relativa o el número de individuos de cada especie bacteriana. La opción "Análisis de componentes principales" se usó para ver las similitudes y diferencias generales entre el muestreo de la gónada, el intestino delgado e intestino grueso, y entre la fracción cultivable y la directa (D) de cada tejido (Chaiyapechara et al., 2012; Pérez et al., 2014, Boon, 2002). Esta distribución de agrupamiento fue verificada por el análisis llevado a cabo con las dos réplicas de gel. Finalmente, el índice de Shannon (H`) que refleja la diversidad de la comunidad bacteriana de ID, IG y G se calculó siguiendo la fórmula: H`=- SPi ln Pi, donde Pi se define como (ni ⁄N), ni es cada banda presente, y N es la suma de todas las bandas (Sun et al., 2011).
Las bandas TTGE con patrones de migración únicos según la escalera de DNA GenRulerTM 100bp (Thermo Fisher Scientific) y las bandas de mayor intensidad en cada TTGE, se escindieron, y el ADN se eluyó siguiendo el método de molienda húmeda descrito por Sambrook y Russell (2002). Luego se tomaron 5 μl de la elución final y se volvieron a amplificar con los iniciadores 341F sin terminación de GC y 907R. Los productos reamplificados de la fracción C se verificaron mediante electroforesis en gel de agarosa al 1%. Los productos amplificados de la fracción D se purificaron utilizando el kit de purificación de productos de PCR QIAquick (QIAGEN CA, EE. UU.) y se clonaron en células competentes de E. coli DH5α usando un kit de clonación Clone JETTM (Thermo Scientific, EE.UU.). La ligadura se realizó siguiendo las indicaciones del fabricante y se completó la transformación según lo descrito por Sambrook y Russell (2002). El inserto se amplificó siguiendo las recomendaciones del kit de purificación QIAquick (QIAGEN CA, EE. UU.) con los iniciadores pJET1.1 y pJET1.2. Los amplicones de D y C se secuenciaron en un Analizador Genético ABI Prism 3100 (Applied Biosystems, CA, EE. UU.).
Las secuencias de DNAr 16S obtenidas de ambas fracciones se editaron usando BioEdit® (Hall, 1999) y se determinó la presencia de quimeras con el programa en línea DECIPHER v2.0 descrito por Wright et al. (2012). Las secuencias editadas se compararon luego con las secuencias de referencia del Centro Nacional de Información Biotecnológica (NCBI, por sus siglas en inglés) y del Proyecto de Base de Datos Ribosomal (RDP, por sus siglas en inglés). Para determinar la afiliación filogenética de los aislamientos microbianos y las bandas obtenidas, se realizaron búsquedas de similitud y análisis filogenéticos utilizando los programas BLASTN y MEGA 7 (Tamura et al., 2007). El análisis filogenético se realizó utilizando el método del vecino más cercano (Saitou y Nei, 1987) con 1000 replicaciones bootstrap. Se enraizó con una secuencia del gen RNAr 16S de Crenarchaeote simbionte de Axinella sp., con el fin de mejorar la topología. Las secuencias obtenidas en este estudio se depositaron en el GenBank con los números de acceso KX891431-KX891450 y KX886796-KAX886797.
RESULTADOS
Recuento bacteriano
La Tabla 2 presenta la abundancia bacteriana promedio de la fracción cultivable en los compartimentos del tracto digestivo y de la gónada en cada medio, expresándose como UFC por gramo de tejido. Los promedios de la abundancia de bacterias que se obtuvieron de la gónada cultivada tanto en agar marino como en TCBS fueron 1,21x107 y 2,83x107 UFC por gramo de tejido, respectivamente. Los recuentos en agar TCBS indicaron una abundancia similar en ambos compartimentos digestivos (1,01x107 UFC por gramo de tejido), mientras que, por el contrario, los recuentos de las bacterias del intestino delgado cultivadas en agar marino revelaron una concentración de bacterias heterotróficas cinco veces superior a la del intestino grueso.
Tabla 2 Abundancia bacteriana promedio (UFC/g) de la gónada, el intestino delgado e intestino grueso en cultivos de agar marino y TCBS.
| Tejido Tissue | Agar marino (UFC/g) Marine Agar (CFU/g) | Agar TCBS (UFC/g) Agar TCBS (CFU/g)T |
|---|---|---|
| Intestino delgado Foregut | 5.57*107 | 1.01*107 |
| Intestino grueso Hindgut | 1.10*107 | 1.00*107 |
| Gónada Gonad | 1.21*107 | 2.83*107 |
Análisis de los perfiles de PCR-TTGE
La estructura de la comunidad bacteriana asociada con los tejidos de la gónada e intestinales obtenida en la fracción cultivable "C" y la fracción directa "D" se evaluó por medio del perfil TTGE de los fragmentos del gen RNAr 16S (Figura 2A y Figura 2B). Los resultados de los perfiles de bandas, evaluados con el programa GelCompar II, revelaron la presencia de varios patrones de migración; estos perfiles revelaron diferencias entre las comunidades bacterianas asociadas a cada tejido y las muestras de la fracción "D" presentaron patrones más complejos y bandas más diversas en comparación con la fracción cultivable "C".

Figura 2 Perfil TTGE de la región V3-V6 del gen RNAr 16S de las fracciones cultivable y directa de la gónada (A), y del intestino delgado e intestino grueso (B). Los rótulos encima de la imagen indican el origen de las muestras. "G" para la gónada, "ID" para el intestino delgado e "IG" para el intestino grueso, "P" para el pool e "I" para el individuo. Los carriles marcados con "L" corresponden a marcadores de 100 pb utilizados como referencia. Los códigos sobre cada carril indican las bandas seleccionadas para la secuenciación.
A partir de este análisis en gel (Figura 2), se seleccionaron 105 bandas de ADN (69 de intestino y 36 de la gónada) únicas y comunes en función de la intensidad de la banda. Con respecto a la gónada de Lobatus gigas, la banda G38 mostró una población predominante asociada con las muestras individuales y combinadas de la fracción directa. La fracción cultivable se caracterizó por la presencia de las bandas G1 y G8 en tres muestras combinadas y cuatro muestras individuales (Figura 2A). Los resultados de las secuencias de bandas de ADN se presentan en la Tabla 4.
Los perfiles genéticos y el análisis de agrupamiento realizados a partir de la fracción directa y cultivable de la gónada revelaron dos grupos diferentes según la fracción, con una similitud del 22,2% para el grupo A y del 27,9% para el grupo B (Figura 3A). Se formaron dos subgrupos en el grupo A a partir de las muestras de la fracción cultivable, una de ellas con una similitud superior al 60% y la otra con un nivel de similitud del 50%. El grupo B contenía las muestras de las fracciones directas (Figura 3A).

Figura 3 Análisis de agrupamiento de los perfiles de TTGE de la fracción cultivable "C" y fracción directa "D" de las muestras individuales "I" y combinadas "P" de la gónada "G" (A), y del Intestino delgado "ID" e Intestino grueso "IG" (B) de L. gigas. El análisis de agrupamiento se realizó con el programa Gel compare II® utilizando el coeficiente de Dice y el método de agrupación de pares no ponderados con medias aritméticas (UPGMA) (Mohammadi and Prasanna, 2003). Las letras de la A a la F y los números representan los grupos y subgrupos generados, respectivamente.
Por otro lado, los perfiles genéticos obtenidos con el método UPGMA para el análisis de agrupamiento del intestino delgado e intestino grueso mostraron seis grupos A, B, C, D, E y F con porcentajes de similitud de 20.7, 45, 38.9, 33.1 y 44.6%, respectivamente (Figura 3B). El grupo A está compuesto por muestras del intestino delgado e intestino grueso, tanto de la fracción cultivable como directa; sin embargo, también se formaron dos subgrupos de acuerdo a la fracción de la que procedían las muestras (Figura 3B, subgrupo 1 y 2). El grupo B agrupó dos muestras individuales (DIDI2 y DIDI16) y una combinada (DIDP5) de la fracción directa del intestino delgado, con un porcentaje de similitud del 45%. El grupo C contenía todas las muestras de la fracción directa del intestino grueso, con un porcentaje de similitud superior al 50%. En el grupo D, se combinaron las muestras de la fracción cultivable, con porcentajes de similitud de 38.9% y 44.6% respectivamente, y se observó un alto porcentaje de similitud en la población bacteriana del intestino delgado. Además, las muestras de las fracciones directas se combinaron principalmente en los grupos B, C y E, con una similitud de 45, 50 y 33.1%, respectivamente. El grupo E contenía muestras del intestino delgado e intestino grueso de las fracciones directas, con niveles de similitud superiores al 33.1%.
Las variaciones en las comunidades bacterianas presentes en los diferentes tejidos (G, ID e IG) observadas en los análisis UPGMA se verificaron por medio de un análisis de componentes principales (PCA) (Figura 4).

Figura 4 (A) Análisis de componentes principales (PCA) de muestras individuales y combinadas de la gónada "G" obtenidos a partir de la fracción cultivable "C" y la fracción directa "D". (B) PCA de muestras de la gónada "G" obtenidas de la fracción cultivable "C" y la fracción directa "D". (C) PCA para evaluar las diferencias entre las comunidades bacterianas asociadas con el intestino delgado "ID" e intestino grueso "IG" de Lobatus gigas. (D). (PCA de los compartimentos digestivos obtenidos de la fracción cultivable "C" y la fracción directa "D".
De acuerdo con la matriz binaria de datos sobre la presencia (1) y ausencia (0) de bandas, se detectaron 36 bandas en los geles de la gónada. Se calculó una matriz binaria de presencia/ausencia de bandas, aplicando el coeficiente de similitud de Dice, y se utilizó para realizar un análisis de similitud (ANOSIM por sus siglas en inglés) basado en el coeficiente de Bray-Curtis, que permite realizar pruebas de significancia de los grupos de datos (Hammer et al., 2001). Se encontraron diferencias significativas entre las muestras asociadas con la fracción cultivable “C” y la fracción directa “D” [ R=0.868 (Bray Curtis) y (Jaccard), (P <0,004 en ambos análisis)]. Además, se demostraron las diferencias entre la composición de la comunidad bacteriana de la fracción cultivable de las muestras individuales y la de las combinadas (Figura 4 A). El Indice de Shannon (H`) mostró una diferencia entre las comunidades bacterianas asociadas con la fracción cultivable "C" y la fracción directa "D" (Tabla 3). Además, el análisis mostró que las poblaciones bacterianas asociadas con las muestras individuales de tejido de la gónada están menos relacionadas que las obtenidas a partir de las muestras combinadas de tejido de la gónada, siendo cierto para ambas fracciones.
Tabla 3 Índice de Shannon (H`) calculado a partir de los perfiles de TTGE de la región V3-V6 del gen RNAr 16S para cada tejido y cada fracción.
| Índice de Shannon (H`) Shannon Index (H`) | |||
|---|---|---|---|
| Tejidos Tissues | Fracción C C Fraction | Fracción D D Fraction | Fracciones C y D C and D Fractions |
| Intestino delgado (ID) / Foregut (F) | 1.4 | 2.5 | 2.5 |
| Intestino grueso (IG) / Hindgut (H) | 1.7 | 1.7 | 2.4 |
| ID e IG / F and H | 2.2 | 2.7 | 2.9 |
| Gónada / Gonad | 1.6 | 1.9 | 2.3 |
Además, se determinaron los resultados del análisis de 69 bandas en los geles del tejido intestinal. El PCA observado en la Figura 4C y el análisis de similitud (ANOSIM) mostraron diferencias significativas en la comunidad bacteriana asociada con el intestino delgado (ID) e intestino grueso (IG) [R=0.1406 y (Bray Curtis) y (Jaccard), (P <0.0018 en ambos análisis)]. Las diferencias entre las comunidades bacterianas relacionadas con las fracciones cultivable y directa de cada compartimento se observan en el PCA (Figura 4D). Se encontraron diferencias significativas entre las muestras asociadas con la fracción cultivable “C” y la fracción directa “D” del intestino delgado (ID) [R=0.3479 (Jaccard) y R=0.3479 (Bray-Curtis), (P <0.0024 en ambos análisis)], y el intestino grueso (IG) [R=0.4643 (Jaccard) y R=0.4643 (Bray Curtis), (P <0.0003)].
Los valores de H` calculados a partir de G, ID e IG (teniendo en cuenta las fracciones C y D) revelaron que el intestino delgado tiene la comunidad bacteriana más diversa, mientras que la gónada tiene la menos diversa. Cuando se calculó H' para cada tejido y para C y D por separado, fue posible observar que la diversidad es mayor en la fracción D que en la fracción C, excepto en el tejido del intestino grueso que mantuvo la diversidad en las dos fracciones (Tabla 3).
La secuenciación y el análisis filogenético de las bandas (≈400 pb) obtenidas del gel TTGE revelaron varios grupos filogenéticos diferentes para cada tejido analizado (gónada, intestino delgado e intestino grueso) de acuerdo a su similitud con las secuencias del gen RNAr 16S en bases de datos públicas (Tabla 4, Figura 5). Estos pertenecían a Alphaproteobacteria, Gammaproteobacteria y Bacilli (Tabla 4, Figura 5A) para la gónada, y la especie más común fue Ralstonia pikettii, que se observó tanto en la fracción cultivable como en la directa (Figura 4A). En los compartimentos digestivos, los resultados de las secuencias parciales del gen 16S rRNA revelaron una gran similitud con las bacterias pertenecientes a Alphaproteobacteria (12.5%), Betaproteobacteria (12.5%), Gammaproteobacteria (12.5%), Bacilli (31.25%), Clostridia (6.25%), Actinobacteria (6.25%), Mollicutes (6.25%) y Deinococci (6.25%), y bacterias no clasificadas (Tabla 4, Figura 5B).
Tabla 4 Afiliaciones taxonómicas de las secuencias parciales de ADN obtenidas de los perfiles TTGE de la gónada, intestino delgado e intestino grueso de L. gigas.
| Origen Origin | Banda TTGE TTGE Band | Nº de acceso Accession N° | Porcentaje de similitud (BlastN) Percent Similarity (BlastN) | Filo Affiliation Phylum | Clase Class | Secuencia más cercana Closest sequence | |||
|---|---|---|---|---|---|---|---|---|---|
| DIDI6 / DFI6 | 4F | KX891442 | 93% | α-Proteobacteria | Oceanicola marinus. NR_043969.1 | ||||
| DIDP1 / DFP1 | 9F | KX891443 | 98% | β-Proteobacteria | Undibacterium oligocarboniphilum. NR_117348.1 | ||||
| DIDI3 / DFI3 | 3F | KX891440 | 97% | Proteobacteria | |||||
| DIGP3 / DPH3 | Clon 38H | KX891439 | 98% | ||||||
| CIGP5 / CHP5 | AH | KX891445 | 99% | γ-Proteobacteria | Pseudomonas azotoformans. NR_113600.1 | ||||
| DIGI16 / DHI16 | Clon 31H | KX891438 | 99% | Pantoea stewartii. NR_104928.1 | |||||
| DIDP3 / DFP3 | Clon 18F | KX891432 | 95% | Firmicutes | Clostridia | Clostridium straminisolvens. NR_024829.1 | |||
| DIDP4 / DFP4 | 21F | KX891442 | 100% | Bacilli | Streptococcus sanguinis. NR_113260.1 | ||||
| DIGI6 / DHI6 | 29H | KX891431 | 100% | Streptococcus sanguinis. NR_074974.1 | |||||
| DIGI3 / DHI3 | Clon 28H | KX891436 | 100% | Streptococcus mitis. NR_116207.1 | |||||
| CIDI2 / CFI2 | ZF | KX891444 | 96% | Anoxibacillus amylolyticus. NR_042225.1 | |||||
| DIDI2 / DFI2 | 2F | KX891437 | 100% | Bacillus cereus. NR_074540.1 | |||||
| DIGI2 / DHI2 | Clon 24H | KX891434 | 99% | Actinobacteria | Actinobacteria | Propionibacterium acnes. NR_040847.1 | |||
| 26H | KX891435 | 88% | Deinococcus- Thermus | Deinococci | Deinococcus geothermalis. NR_074342.1 | ||||
| DIGP5 / DHP5 | 42H | KX891441 | 88% | Tenericutes | Mollicutes | Mycoplasma neophronis. NR_108494.1 | |||
| DIDP1 / DFP1 | Clon 10F | Secuencia de la quimera | 93% | Spirochaetae | Spirochaetia | Spirochaeta litoralis. NR_104732.1 | |||
| CGI4 / CGI4 | G1 | KX891446 | 96% | Ralstonia pickettii. NR_043152.1 | |||||
| 96% | Ralstonia pickettii. NR_102967.1 | ||||||||
| G8 | KX891447 | 96% | β-Proteobacteria | Ralstonia pickettii. NR_043152.1 | |||||
| 96% | Ralstonia mannitolilytica. NR_025385.1 | ||||||||
| DGP5 / DGP5 | Clon G38 | KX886796 | 99% | Proteobacteria | Ralstonia pickettii. NR_043152.1 | ||||
| 98% | Ralstonia mannitolilytica. NR_025385.1 | ||||||||
| CGP5 / CGP5 | G22 | KX891450 | 95% | α-Proteobacteria | Roseomonas aquatica. NR_042501.1 | ||||
| 94% | Roseomonas vinacea. NR_044191.1 | ||||||||
| CGP4 / CGP4 | G21 | KX886797 | 83% | Firmicutes | Bacilli | Bacillus gotheilli. NR_108491.1 | |||
| 83% | Bacillus foraminis. NR_042274.1 | ||||||||
| DGP2 / DGP2 | G32 | KX891449 | 94% | Bacillus lichenoformis. NR_074923.1 | |||||
| 94% | Bacillus shackletonii. NR_025373.1 | ||||||||

DISCUSIÓN
Estudios recientes han caracterizado la microbiota de L. gigas del Caribe colombiano, con el objetivo de estudiar la diversidad bacteriana asociada con los caracoles silvestres y cautivos (Acosta et al., 2009; Rodriguez et al., 2011; Pérez et al., 2014). En este estudio, realizamos una de las primeras investigaciones exploratorias en diferentes tejidos para examinar la comunidad bacteriana dominante en L. gigas. Analizamos la composición bacteriana de la gónada y el intestino (delgado y grueso) del caracol rosado de la Reserva de la Biosfera de Seaflower mediante el uso de diferentes métodos microbiológicos y moleculares. Cuando comparamos las bacterias cultivadas, el enfoque molecular basado en el análisis del extracto de ADN directamente obtenido de la muestra parece ser una estrategia complementaria para determinar los componentes principales en la comunidad microbiana del caracol rosado.
El análisis realizado utilizando las técnicas de cultivo convencionales, tales como el recuento de UFC en agar marino y TCBS, nos permitió establecer la presencia de bacterias heterotróficas y bacterias pertenecientes a la familia Vibrionacea en la gónada y en los compartimentos digestivos del caracol rosado. El recuento en agar TCBS sugiere una abundancia de vibrios con un total de 2.83 x 107 UFC por gramo de tejido gonadal y 1.01 x 107 UFC por gramo de tejido para los compartimentos digestivos analizados; sin embargo, hasta la fecha no hay estudios que hayan descrito la microbiota gonadal del caracol rosado. Previamente, Avendaño-Herrera et al. (2001) reportaron una carga de bacterias heterotróficas en la gónada del bivalvo Argopecten purpuratus cautivo; obtuvieron un recuento de 4 x 104 UFC por gramo y un recuento de Vibrionacea de 3 x 102 UFC por gramo. Nuestros resultados contrastan con los descritos por Avendaño-Herrera et al. (2001) lo que podría deberse a las condiciones de cautividad bajo las cuales se realizaron los estudios de A. purpuratus. La afinidad de los vibrios por el tejido de la gónada se ha descrito previamente en el molusco Crassostrea gigas y se ha relacionado con los episodios de mortalidad de esta especie (De Decker et al., 2011). Se necesitan estudios destinados a establecer la identidad de los vibrios asociados con la gónada, el intestino delgado e intestino grueso y sus posibles efectos sobre el desarrollo de los moluscos. Además, las bacterias pertenecientes a la familia Enterobacteriaceae y los géneros Aeromonas y Pseudomonas pueden desarrollarse en el medio TCBS, y podrían interferir con los recuentos bacterianos, generando una sobreestimación en la abundancia total de vibrios presentes en el tejido analizado.
El análisis de los perfiles de TTGE mostró comunidades bacterianas similares entre las muestras individuales y las combinadas en el tejido de la gónada (Figura 2A), con predominio de dos o tres bandas por perfil. Estos resultados podrían estar relacionados con las bacterias autóctonas de este tejido del molusco. Las diferencias en los perfiles de bandas observados entre las muestras individuales y combinadas de la gónada, el intestino delgado e intestino grueso se verificaron con un PCA. El análisis sugiere con fuerza que hay una mayor diferencia en las poblaciones bacterianas asociadas con las muestras individuales que las asociadas con las muestras combinadas de ambas fracciones (C y D) (Figura 3). Se han reportado resultados similares en diferentes estudios con organismos marinos. En estos estudios, se observó una variación individual incluso entre los peces criados en condiciones ambientales similares, con antecedentes genéticos similares y alimentados con la misma dieta, lo que pone de relieve la influencia del hospedador en la diversidad bacteriana (Spor et al., 2011; McKnite et al., 2012; Navarrete et al., 2012). Además de esto, los perfiles de las muestras combinadas fueron similares en algunas muestras, lo que sugiere la presencia de una población bacteriana dominante en estos tejidos. Sin embargo, estos resultados podrían llevar a conclusiones sesgadas, debido a la posibilidad de que las bacterias dominantes presentes en un individuo en particular puedan interpretarse como bacterias comunes para todos los individuos analizados en la muestra (Reveco et al., 2014).
Por otro lado, los resultados revelaron la presencia de una comunidad más compleja y diversa asociada con el intestino delgado del caracol rosado en comparación con el intestino grueso. También se observaron cambios importantes en la estructura de la comunidad bacteriana entre la fracción cultivable (C) y la fracción directa (D). Se encontraron diferencias significativas entre ambos compartimentos digestivos con un porcentaje promedio de disimilitud del 94.41%. Estos resultados se verificaron con el análisis de componentes principales presentado en la Figura 4C, el cual sugiere que el intestino delgado e intestino grueso han sido colonizados por diferentes comunidades bacterianas; este hecho fue respaldado por el índice de diversidad (H`), que resultó ser mayor en el intestino delgado que en el intestino grueso (Tabla 3). Estas diferencias podrían estar asociadas con los procesos metabólicos que tienen lugar específicamente en cada compartimento. Los estudios en peces sugieren que la variación en la diversidad microbiana entre los compartimentos digestivos podría deberse a diferencias en el pH y a la actividad de la proteasa en la luz intestinal (Yu et al., 2007; Yúfera and Darías, 2007). Además, se han reportado diferencias en el pH del estómago, ciegos pilóricos e intestino del pez Epinephelus coioides (Yu et al., 2007). Estos parámetros fisicoquímicos podrían estar asociados con los cambios observados en la microbiota de los diferentes compartimentos digestivos, que podrían estar relacionados con las funciones específicas de cada intestino y las condiciones ecológicas del hábitat donde se alimenta L. gigas.
Según el análisis filogenético, las secuencias de la gónada indicaron que la comunidad bacteriana había sido colonizada principalmente por bacterias pertenecientes al género Ralstonia, que estaban representadas por varias bandas TTGE en las fracciones directa y cultivable de las muestras individuales y combinadas (Figura 2A, Tabla 4). Este hallazgo sugiere que la gónada podría ser un sitio de simbiosis bacteriana en el caracol rosado. Aunque se sabe poco sobre la microbiota de la gónada en organismos marinos y su interacción con el hospedador, estudios previos del microbioma en mamíferos han demostrado que existe una simbiosis entre mamíferos y bacterias en el tracto reproductor y que esta interacción puede ayudar a defender al hospedador contra las infecciones y mejorar su salud reproductiva (Miller et al., 2017; Kindinger et al., 2017).
En el caso del intestino delgado e intestino grueso, el análisis filogenético reveló diferencias entre los dos compartimentos (Tabla 4 y Figura 5B). Las especies encontradas en ambos compartimentos pertenecen al phylum Firmicutes y Actinobacteria, mientras que los filos Proteobacteria, Deinococcus - Thermus, y Tenericutes solo se encontraron en el intestino grueso del caracol rosado. Estos resultados son consistentes con los reportados en muestras intestinales del caracol rosado de las Islas del Rosario (Acosta et al., 2009; Pérez et al., 2014) y en otros organismos marinos (Sun et al., 2011; Trabal et al., 2012; Chaiyapechara et al., 2012). Aunque existen diferencias en las metodologías empleadas, los resultados están relacionados con la alta diversidad bacteriana asociada con este molusco.
Las bacterias de los géneros Bacillus y Propionibacterium han sido caracterizadas como probióticos en peces y crustáceos marinos (Nakagawa et al., 2007; Lan et al et al., 2007; Cousin et al., 2010). Más específicamente, se ha establecido que el género Bacillus actúa como un agente biológico contra Aeromonas hydrophila (Lalloo et al., 2007, 2008) y los miembros de este género producen una amplia gama de compuestos antagónicos que se han encontrado en el tracto digestivo de crustáceos y peces (Balcázar et al., 2006). La presencia de Pseudomonas en el intestino delgado del caracol rosado coincide con los resultados reportados en otros moluscos y en diferentes organismos marinos como crustáceos y peces (Sfanos et al., 2005; Romero and Navarrete, 2006; Trabal et al., 2012). Es importante observar que los miembros de este género, aislados de los organismos marinos, han sido reconocidos por su actividad antibiótica frente a un amplio espectro de microorganismos (Gram, 1993; Jayatilake et al., 1996; Zheng et al., 2005). Estos resultados sugieren que la presencia de Pseudomonas libanensis podría estar relacionada con efectos beneficiosos y contribuir a la supervivencia de L. gigas al defenderle de posibles patógenos que puedan afectar a su salud y desarrollo. Mientras tanto, el género Streptococcus (Tabla 4 y Figura 5B) ha sido asociado con varias enfermedades en peces y crustáceos, e identificado como el agente causante de la enfermedad del desarrollo lento del crustáceo Carcinus mediterraneus (Pappalardo y Boemare, 1982). Los géneros Undibacterium y Oceanicola encontrados en el intestino delgado e intestino grueso del caracol rosado (Tabla 4) también se han reportado recientemente en el intestino de las muestras del crustáceo silvestre Penaeus monodon (Rungrassamee et al., 2014). Resulta llamativo que Oceanicola marinus sea una especie común en ambientes marinos (Lin et al., 2007; Chunyan et al., 2012).
Se observaron diferencias entre los resultados obtenidos del análisis de las fracciones cultivable y directa en la gónada, el intestino delgado e intestino grueso del caracol rosado. Es posible que estas diferencias se deban a los métodos dependientes de cultivo-los cuales favorecen el crecimiento de bacterias capaces de crecer en medios sintéticos-independientemente de si estas bacterias son dominantes en la muestra. Se ha demostrado que estas bacterias representan sólo el 1% de la población total, lo que subestima la diversidad de las poblaciones microbianas existentes (Hansen y Olafsen, 1999; Suau et al., 1999). Otros estudios basados en métodos independientes de cultivo han demostrado una imagen menos sesgada de la población bacteriana presente en las muestras ambientales que los métodos dependientes de cultivo (Amann et al., 1995). No obstante, es necesario tener en cuenta que las técnicas basadas en genes ribosomales pueden verse afectadas por las bajas concentraciones de ADN en diferentes especies o la presencia de altas concentraciones de ADN que compiten durante las reacciones de la PCR (Ogier et al., 2002), lo que puede afectar a la detección de algunas especies en la comunidad (Kisand y Wikner, 2003). Esta es la razón por la cual es necesario desarrollar ambos enfoques (dependientes de cultivo e independientes de cultivo) en el estudio de la microbiota a partir de las muestras naturales, con el fin de recoger la mayor cantidad de información posible sobre las especies bacterianas presentes en las muestras analizadas (Kisand y Wikner, 2003).
Esta investigación determinó la comunidad bacteriana asociada con la gónada y el intestino delgado e intestino grueso de L. gigas. Con respecto a la gónada, los resultados sugieren que este órgano tiene una microbiota estable dominada principalmente por miembros del género Ralstonia que podrían ser endosimbiontes de este tejido. Además, es necesario verificar la presencia de vibrios y determinar su efecto sobre la gónada. Aún se requieren estudios más detallados para determinar el papel que desempeña esta comunidad bacteriana en la salud y el ciclo reproductivo de L. gigas. En el caso del intestino delgado e intestino grueso, el análisis filogenético reveló diferencias entre los dos compartimentos. Sin embargo, se necesitan más estudios para establecer el efecto de estas comunidades en los diferentes compartimentos del caracol rosado. En general, los hallazgos que describimos podrían usarse para el desarrollo de estrategias para lograr aumentar la nutrición, la prevención de patógenos y contribuir a la conservación de esta especie vulnerable de la Reserva de la Biosfera de Seaflower en el Caribe colombiano.

