











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCCIÓN
En los últimos años se ha intensificado la búsqueda de hongos capaces de modificar la pared celular con el fin de encontrar mejores sistemas de degradación de la materia orgánica (Dhouib et al., 2005). La familia Xylariaceae pertenece a un grupo de hongos ascomicetes relacionados con la pudrición blanda y son un componente importante de la microbiota en bosques tropicales donde actúa principalmente como hongo descomponedor (Floudas et al., 2012). Su habilidad está relacionada con la producción de enzimas extracelulares como las hidrolasas versátiles y lacasas que permiten degradar material orgánico (Castaño et al., 2015). Por esta razón el estudio de la expresión de genes que codifican enzimas lignocelulolíticas ha generado un gran interés debido a su uso en diferentes aplicaciones biotecnológicas (Kunamneni et al., 2007).
El análisis de expresión génica es piedra angular en la biología molecular moderna y contribuye a un mejor entendimiento de la genética y la regulación transcripcional (Liu et al., 2012). Dentro de las metodologías para el análisis de expresión de genes se encuentra la reacción en cadena de la polimerasa con transcriptasa reversa (RT-PCR) y la PCR cuantitativa en tiempo real (RT-qPCR), ambas técnicas permiten la detección de mARNs transcritos por genes específicos y son ampliamente utilizadas debido a su alta precisión, sensibilidad, especificidad y reproducibilidad (Walker, 2002).
La obtención de ARN de calidad constituye la base para el desarrollo de diferentes técnicas moleculares, de lo contrario podría comprometer seriamente los resultados de procedimientos posteriores a la extracción, los cuales requieren intensa labor, tiempo adicional y altos costos. Extraer ARN no siempre es sencillo, esta molécula es menos estable que el ADN y la presencia de contaminantes como ARNsas, proteínas, polisacáridos y ADN genómico pueden desafiar su obtención (Zakaria et al., 2013). Adicionalmente, se ha reportado que la presencia de estos contaminantes puede interferir con la amplificación de los ácidos nucleicos (Vermeulen et al., 2011).
En el caso particular de hongos, la obtención de ARN de baja calidad es frecuente debido a la presencia de paredes celulares o estructuras como las hifas y conidias que no son degradables fácilmente (Melo et al., 2006). Adicionalmente, la presencia de alto contenido de fenoles y polisacáridos genera la necesidad de modificar los protocolos de extracción (Vasanthaiah et al., 2008; Kumar, 2012). En la actualidad, existen diferentes metodologías de ruptura y homogeneización de tejidos a través del uso de nitrógeno líquido, arena, esferas o liofilización de micelio (Burden, 2008), sin embargo, independientemente de la metodología usada, el riesgo de rehidratación de muestras y activación de ARNasas está siempre latente. A la fecha, se han reportado diferentes procesos de extracción de ARN entre las cuales se encuentran el uso de compuestos fenólicos, triazoles, sodio dodecil sulfato (SDS), cloruro de litio, detergentes como el Brumoro de hexadeciltrimetilamonio (CTAB) y ahora con mayor frecuencia con paquetes o kits comerciales de extracción (Rojas et al., 2011; Lever et al., 2015). No obstante, la calidad y rendimiento del ARN extraído puede variar entre las metodologías mencionadas y la especie biológica de estudio (Fleige & Pfaffl, 2006).
Los genes que codifican proteínas esenciales para la vida o ‘housekeeping genes’ (HKG) son útiles como genes de referencia para estandarizar técnicas de expresión génica, ya que estos se transcriben de manera estable durante el ciclo de vida de la célula y en diferentes estados de desarrollo (Giménez, 2011). Por lo tanto, permiten corregir variaciones tales como diferentes cantidades de ARN inicial y eficiencias durante la transcripción reversa (Udvardi et al., 2008). No obstante, es recomendable evaluar diferentes cebadores, para ampliar el rango de opciones y elegir el que más se ajuste a las condiciones experimentales (Hruz, 2011). Para la familia Xylariaceae se han diseñado cebadores para el gen β-tubulina y actina utilizados en estudios filogenéticos y de expresión génica, respectivamente (Chen et al., 2013).
Se ha determinado que al usar metodologías con alta reproducibilidad y sensibilidad, como RNAseq, se pueden generar cambios en la expresión génica dependiendo de la metodología de extracción de ARN utilizada (Sultan et al., 2014). Por lo tanto, esta investigación tuvo como objetivo evaluar diferentes metodologías de homogeneización (nitrógeno líquido y liofilización) y extracción de ARN (Trizol, CTAB y RNeasy mini kit) a partir de micelio del hongo ascomicete Xylaria sp. Esto con el fin de estandarizar una metodología que permita la obtención de ARN de alta calidad y rendimiento. Adicionalmente, como modelo de estudio y validación ser realizó una RT-PCR, usando cebadores degenerados para amplificar un fragmento del gen de la β-tubulina. Estos fueron diseñados mediante análisis múltiple de secuencias obtenidas de hongos ascomicetos.
MATERIALES Y MÉTODOS
Aislamiento fúngico y preservación
Los aislamientos del hongo Xylaria sp. (Ascomicota: Xylariaceae) fueron obtenidos en el laboratorio de Agrobiotecnología de la Facultad de Ciencias Agrarias (FCA) de la Universidad Nacional de Colombia (UNAL), sede Bogotá. Este hongo fue seleccionado previamente por su alta actividad lacasa y preservado en medio agar-salvado de trigo a 4°C según Castaño et al, (2015). Para evaluar la especificidad de los cebadores de referencia diseñados, además del ADN extraído a partir del hongo Xylaria sp., se utilizó el ADN de otros hongos actinomicetos (Sclerotinia sp., Fusarium solani, Penicillium sp., Fusarium oxysporum) y basidiomicetos (Hymenogloea sp., Pholliota sp., Agrocybe sp.) obtenidos previamiente en el laboratorio de Agrobiotecnología (Crespo et al., 2014).
Medios de cultivo
Los hongos se sub-cultivaron en 30 ml de medio de cultivo líquido Mandels modificado de acuerdo a lo reportado por Moya & Torres (2012). En todos los casos, los medios de cultivo fueron inoculados con tres discos de 5 mm de diámetro tomados de la periferia del hongo e incubados a 26 °C y con agitación de 150 rpm durante 6 días de acuerdo a lo reportado por Castaño et al, (2015).
Extracción de ARN total a partir de micelio
Para el proceso de extracción de ARN se partió de micelio homogenizado mediante maceración con nitrógeno líquido o liofilización (Christ Alpha 1-4LD) por 48 horas. Todas las soluciones y materiales se esterilizaron tres veces previamente a su utilización a 121ºC por 20 min. Tubos eppendorf de 2 ml fueron alicuotados con aproximadamente 400 mg de tejido fúngico homogenizado. Se evaluaron tres metodologías de extracción de ARN: Trizol® reagent (Life Technologies), protocolo CTAB con precipitación con LiCl2 (Chang et al., 1993) y RNeasy Mini Kit® (Qiagen), de acuerdo a las instrucciones del proveedor. En cada caso, se realizaron tres repeticiones por muestra.
Análisis y cuantificación de ARN
Las soluciones de ARN obtenidas se cuantificaron con un espectrofotómetro Nanodrop ND-2000 (Thermo scientific). Se determinó la pureza del ARN en cada muestra por medio de relaciones de absorbancia A260/A280 y A260/A230 y el rendimiento fue calculado como la cantidad de ARN obtenido (ng) sobre el peso de micelio utilizado para la extracción (µg), las extracciones se realizaron por triplicado. Los resultados de pureza y rendimiento fueron analizados mediante ANOVA y pruebas de comparación de medias de Tukey (α<0.01) siguiendo un diseño factorial completamente al azar, posterior a la revisión de supuestos de normalidad (Lilliefors: p>0.05) y homogeneidad de varianzas (Fligner-Killeen: p>0.05). Los datos obtenidos fueron procesados con el programa estadístico R 3.4.0. Además, se constató y documento la integridad del ARN mediante visualización directa en gel de electroforesis (agarosa al 1% teñido con Bromuro de etidio, 100V por 15min) mediante un visualizador de imágenes ChemiDoc MP system (BioRad). Los procesos de extracción de RNA se llevaron a cabo en el laboratorio de Agrobiotecnología de la FCA de la UNAL, sede Bogotá y la visualización en el laboratorio de Biología de la Facultad de Ciencias de la UNAL.
Diseño de cebadores de referencia para el gen de la β-tubulina
Se diseñó un juego de cebadores a partir de un alineamiento múltiple de veintitrés secuencias de ADN del gen β-tubulina obtenidas de veintitrés especies de la familia Xylariaceae y diez secuencias adicionales de diversos ascomicetes almacenadas en la base de datos NCBI usando BLAST (accesiones: HM585018, GQ502718, GQ502698, GQ502712, GQ502703, GQ502710, GQ502697, GQ487702, GQ487701, GQ495955, GQ502685, GQ502707, GQ478209, GQ502706, GQ495945, AB625369, GQ502692, GQ478222, JX868543, EF025617, JX868550, GQ478226, EF025606, AY951665, AY951734, GQ478213, GQ470224, GQ470228, EF025607, GQ470220, EF025616, GQ478211, JQ691672. Última fecha de acceso: Marzo 2017). Las secuencias consenso se identificaron a partir de alineamientos múltiples para lo cual se utilizó los programas ClustalX2 y GeneDoc. También se utilizaron los programas Primer3, Oligo Analyzer y mFold para el diseño de cebadores y análisis de las propiedades termodinámicas in silico. Los cebadores fueron diseñados teniendo como referencia los siguientes parámetros: temperatura de disociación (Tm: 55-65°C), contenido de Guaninas y Citosinas (GC%: 50-55%), energía óptima para ruptura de estructuras secundarias (ΔG>-9Kcal/mol), tendencia del primer a formar estructuras secundarias con si mismo (3>Any number) y con su extremo 3` (3 > 3` number) (Dieffenbach et al., 1993; Arif & Ochoa-Corona, 2013).
Evaluación del juego de cebadores
Se realizó la validación y optimización de condiciones para el funcionamiento de los cebadores diseñados según Ochoa-Corona et al. (2007), por medio de la evaluación de un gradiente de temperatura óptima de anillamiento (51-62°C), prueba de sensibilidad (diluciones seriadas a partir de 1ng-1pg de ADN de Xylaria sp.) y finalmente prueba de especificidad con ocho especies de hongos lignoceluloliticos. Las reacciones de PCR fueron llevadas a cabo en 50µl de mezcla de reacción que contenía 500ng de ADN, 0.5U de Taq ADN polimerasa, 0.3mM dNTPs, 1X buffer Tris HCl 0.1M, 2mM MgCl2, 0.2µM de primer BtubF (5’-TTCCARATYACMCACTCGCT-3’) y BtubR (5’-GCCATCATGTTCTTRGGGTC-3’). La amplificación de ADN se realizó bajo las siguientes condiciones: 96 °C por 2 min, seguido de 35 ciclos de 95 °C por 1 min, 55 °C por 1 min, 72 °C por 2 min, con una extensión final de 72 °C por 10 min en un termociclador (Thermal Cycler 170-9701, Bio Rad). Finalmente, 5µl de producto de PCR con 1µl de EZ-visión fue visualizado y fotografiado en un gel (agarosa 1.0%, buffer TBE 0.5X, 90V por 35 min) utilizando el visualizador de imágenes ChemiDoc MP system (Bio Rad). Estos procesos fueron llevados a cabo en el laboratorio de Agrobiotecnología de la FCA de la UNAL.
Condiciones de RT-PCR
La contaminación con ADN en la solución de ARN fue eliminada mediante tratamiento con kit de DNase I (Invitrogen, ThermoFisher scientific®) de acuerdo a las indicaciones del proveedor, seguido por síntesis y amplificación del cADN a partir de 1µg ARN total, usando el kit RT-PCR one step (Qiagen), de acuerdo a las indicaciones del proveedor.
RESULTADOS Y DISCUSION
Homogeneización de micelio y extracción de ARN
Como consecuencia a la susceptibilidad del ARN a ser degradado durante el proceso de extracción, la estandarización de este tipo de procedimientos es fundamental para asegurar la reproducibilidad de protocolos. Generalmente, en los procesos de extracción de ácidos nucleicos se hace uso de maceración con nitrógeno líquido para la homogeneización de tejidos, no obstante, existe riesgo de rehidratación de muestras y con ello activación de ARNasas, lo cual puede ocurrir si el procedimiento no es continuo y rápido. (Wilfinger & Mackey, 2015). Otra forma de homogeneizar el tejido inicial es haciendo uso de la liofilización debido a su facilidad durante la manipulación de muestras, la disrupción uniforme del tejido y sus consistentes resultados en los rendimientos de extracción de ARN (Pearson et al., 2006).
En el caso de Xylaria sp. se encontró que tanto el método de homogenización como el protocolo utilizado, afectan el rendimiento de extracción de ARN (figura 1), encontrando los mayores rendimientos con el uso de liofilización y el protocolo RNeasy (306 ngRNA µg-1). Se detectó que con el uso de tejido liofilizado se obtuvieron rendimientos de extracción más altos (1.7, 3.4, 1.5 veces más) que cuando se realizó con micelio macerado con nitrógeno líquido, con las tres metodologías de extracción de ARN empleadas (Trizol, CTAB y RNeasy mini kit, respectivamente). Este resultado puede estar asociado a la diferencia en peso seco de las muestras como resultado de las diferentes metodologías de homogeneización empleadas. Kumar et al. (2007), reportan de manera similar aumentos en rendimiento de extracción de ARN en Solanum tuberosum (Solanaceae) cuando se utiliza tejido liofilizado (100% peso seco) en contraste con tejido macerado con nitrógeno líquido (tejido con 18% peso seco).

Figura 1 ARN obtenido mediante diferentes metodologías de homogeneización y extracción, 1µg de RNA fue servido en cada pozo. Asteriscos indican los P-valores generados por el análisis de varianza (ns, no significativo; *, <0.05; **, <0.01; ***, <0.001) de los factores: método de homogenización (H), tipo de protocolo de extracción (P) y su interacción (H*P). Valores con la misma letra no son estadísticamente diferentes según pruebas de Tukey (á=0.01; n=3).
No obstante, a pesar de haber obtenido indicadores altos de calidad con la homogenización mediante liofilización (figura 1. A260/280 y A260/230 > 1.8: Manchester, 1996), al observar la integridad del ARN en el gel se constató una ligera degradación por el uso de esta metodología de homogeneización, lo cual sugiere que la evaluación de calidad y pureza por medio de espectrofotometría no corresponde completamente a lo reflejado por el gel de electroforesis. Kasajima et al. (2013), discuten que las estimaciones de calidad y pureza de ácidos nucleicos se hacen de manera más confiable con visualización directa en gel de electroforesis, que con el uso de relaciones A260/280 y A260/230. Los resultados obtenidos concuerdan con Sukumar et al. (1997), donde la utilización de tejido liofilizado de Gossypium hirsutum L. (Malvacea) generó una degradación total de ARN, este tipo de daños en la integridad del ARN pueden posteriormente limitar la eficiencia de reacciónes de RT-PCR (Fleige & Pfaffl, 2006). Por otro lado, aunque la maceración con nitrógeno no fue la metodología que permitió los mayores rendimientos de extracción por peso de muestra usada en comparación al uso de liofilización, se logró extraer ARN no degradado, observándose con claridad las dos bandas correspondientes a las subunidades ribosomales 28 y 18S, respectivamente (figura 1).
Con respecto a las metodologías de extracción se encontró que las extracciones con Trizol y RNeasy mini kit® generaron ARN contaminado con polisacáridos (A260/230<1.8; figura 1). Es frecuente encontrar extracciones de ARN de hongos con baja relación A260/230 debido a contaminación con melanina, la cual también absorbe luz en el espectro 200-400nm (Dorrie et al., 2006). Adicionalmente, algunos autores han reportado baja calidad de ARN por presencia de contaminación con polisacáridos mediante el uso de Trizol en plantas y hongos (Ma & Yang, 2011; Schumann et al., 2013); en forma similar, la contaminación por presencia de azúcares residuales al final de la extracción, puede ocurrir cuando se emplean kits de extracción ya que los carbohidratos de la muestra pueden establecer interacciones hidrofóbicas con la matriz la cual posee los grupos oligo dT necesarios para la captura de las secuencias terminales polyA+ del ARN, (Sánchez et al., 2008).
El uso del protocolo CTAB en Xylaria sp. incrementó la pureza, disminuyendo la presencia de polisacáridos (A260/230: 2.43 y 2.63 con nitrógeno y liofilización respectivamente). Se ha reportado que el uso de agentes como CTAB y LiCl2 son ideales para incrementar la pureza del ARN en organismos con alto contenido de contaminantes (Sánchez et al., 2008: Vasanthaiah et al., 2008), esto sucede debido a que el CTAB se une a los polisacáridos cuando la concentración de sales es alta, removiéndolos de la solución (Clarke, 2009). Adicionalmente, el LiCl2 facilita la precipitación específica del ARN, libre de ADN, proteínas o carbohidratos (Barlow et al., 1963); no obstante, en este trabajo, se observó una ligera degradación del ARN con esta metodología de extracción (figura 1).
Con respecto a Xylaria sp. el uso de homogeneización del micelio con nitrógeno líquido combinado con el protocolo de extracción RNeasy mini kit permitió la extracción de ARN de mejor calidad, pureza y rendimiento (A260/280: 2.18; A260/230: 1.5; rendimiento: 209ngRNA µg-1), además este protocolo requiere de un periodo de procesamiento menor a 30 min en comparación con el uso de las otras metodologías de extracción en donde el proceso requiere de aproximadamente 30 horas, lo cual es recomendable cuando se requiere realizar la extracción de un amplio número de muestras.
Diseño del cebadores de referencia para el gen β-tubulina
Con el fin de evaluar la efectividad de las metodologías de extracción de ARN se diseñó un par de cebadores de referencia para la detección del gen β-tubulina, este codifica una proteína crucial requerida en procesos celulares de eucariotes y está presente en la mayoría de los componentes del citoesqueleto y flagelos eucarióticos (Faguy & Doolittle 1998). El gen que codifica la proteína β-tubulina es un candidato ideal como gen de referencia debido a la alta similitud entre secuencias de toda la taxa eucariota (65-70%) (Einax & Voigt, 2003). El análisis realizado con ClustalX2 permitió la identificación de una región altamente conservada en uno de los exones por lo cual se diseñó un par de cebadores en esa posición (figura 2).

Figura 2 Localización aproximada de los cebadores BtubF y BtubR sobre la secuencia del gen β-Tubulina.
A pesar de los recientes desarrollos en herramientas bioinformáticas, el diseño de cebadores puede ser perfeccionado con base en el análisis termodinámico de las secuencias que permiten predecir la eficiencia del cebador incluso antes de su validación in vitro en reacciones de PCR (Arif & Ochoa-Corona, 2013). Por esta razón, mediante el programa mFold se determinó la tendencia de los cebadores a formar estructuras secundarias y se encontró que la energía óptima (ΔG) es de 1 y 0.7 kcal/mol para los cebadores sentido y antisentido, respectivamente (tabla 1). Este valor hace referencia a la cantidad de energía mínima requerida para romper dichas estructuras, ΔG menores a -9 kcal/mol pueden generar problemas en reacciones de PCR (IDT, 2010), generalmente asociados a alto contenido de regiones GC que requerirían condiciones denaturantes más agresivas como temperaturas de disociación más altas, ocasionando un rápido deterioro de la polimerasa. Por lo tanto, se requiere que los cebadores diseñados tengan un rango mínimo de energía que no limite las condiciones de la reacción PCR.
Tabla 1 Evaluación de propiedades termodinámicas del juego de cebadores β-Tub.

*Valor ∆G calculado usando mfold
***Tendencia oligo a formar estructura secundaria calculado usando Primer 3
****Tendencia del extremo terminal 3´ del oligo a formar dímeros con sí mismo calculado usando Primer 3
El análisis realizado determinó que los cebadores poseen características termodinámicas ajustadas a un margen óptimo que favorece la amplificación de ADN en PCR (tabla 1). Los cebadores diseñados presentan baja probabilidad de formación de estructuras secundarias (ANY< 3; y 3’< 6), adicionalmente poseen otros parámetros óptimos como son: %GC (55%) y diferencia entre Tm de los cebadores (< 2°C). Los cebadores que no cumplen con estas características pueden experimentar dimerización, amplificaciones inespecíficas e incluso falsos positivos (Dieffenbach et al., 1993). El análisis realizado permite predecir que los primer poseen características termodinámicas favorables para una eficaz reacción de PCR (Arif & Ochoa-Corona, 2013).
Los cebadores ΒtubF y ΒtubR fueron validados y optimizados por un gradiente de temperatura de anillamiento en donde la temperatura óptima correspondió a 57 °C (figura 3A); temperaturas menores a 53 °C favorecieron amplificaciones inespecíficas y temperaturas mayores a 61 °C inhibieron la hibridación de los cebadores. Mediante una prueba de sensibilidad se determinó que este par de cebadores poseen un límite de amplificación de hasta 1ng de ADN (figura 3B), otros autores han reportado sensibilidades de detección de 25 ng (Quellhorst & Rulli, 2006), cebadores altamente sensibles tienen la ventaja de permitir la detección el objetivo en muestras con poco ARN. Finalmente, mediante la prueba de especificidad se determinó que en ascomicetes se detecta una única banda, cuyo peso varía entre 500-650pb dependiendo de la especie (figura 3C); esto es ideal ya que múltiples bandas indican generalmente la presencia de inespecificidad (Quellhorst & Rulli, 2006). Los cebadores amplifican productos en diferentes organismos ascomicetes y algunos basidiomicetes, por lo que se concluye que se diseñaron cebadores en una región altamente conservada que permiten su uso en el estudio de genes de los diversos organismos evaluados.
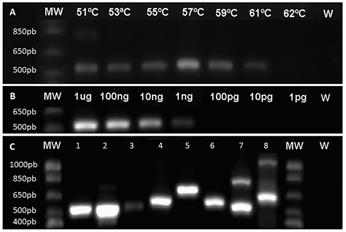
Figura 3 Validación y optimización de los cebadores β-tub mediante: (A) evaluación de gradiente de temperatura de anillamiento, (B) evaluación de sensibilidad y (C) evaluación de especificidad en diferentes ascomicetes (1: Xylaria sp.; 2: Sclerotinia sp.; 3: Fusarium solani; 4: Penicillium sp.; 5: Fusarium oxysporum) y basidiomicetes (6: Hymenogloea sp.; 7: Pholliota sp.; 8: Agrocybe sp.). 1kb marcador ( (MW), Control negativo con agua (W).
RT-PCR
La evaluación de la integridad y pureza del ARN es un factor crítico previo a la validación de datos de expresión génica (Fleige & Pfaffl, 2006; Vermeulen et al., 2011). Con el ARN extraído mediante las diferentes metodologías descritas se evaluó con los cebadores de referencia diseñados y se pudo comprobar que a pesar de la contaminación con proteínas y polisacáridos detectada en varias de las metodologías utilizadas (figura 1), se logró la amplificación exitosa del primer de referencia en todos los tratamientos (figura 4).

Figura 4 Evaluación de la eficiencia de la reacción de RT-PCR a partir de ARN obtenido por diferentes metodologías de homogeneización y extracción. MW: 1kb marcador (, W: control negativo con agua.
Particularmente el ARN obtenido con el protocolo con CTAB generó una banda tenue en comparación con las obtenidas con ARN proveniente de otras metodologías de extracción. Estos resultados sugieren que el ARN obtenido mediante este protocolo posiblemente generó una disminución en la eficiencia de la reacción de RT-PCR. Sánchez et al. (2008), reporta que incluso después de centrifugación, considerables cantidades remanentes de buffer de lisis pueden permanecer atrapadas en los espacios internos del material amorfo precipitado y trazas de inhibidores de detergentes como el SDS podrían tener un efecto sobre la eficiencia de la reacción de PCR (Rossen et al., 1992), generando finalmente bandas tenues en el gel de electroforesis.
CONCLUSIONES
El uso de tejido liofilizado generó los mayores rendimientos de ARN durante el proceso de extracción por masa de micelio inicial, comparado con la maceración con nitrógeno líquido; sin embargo, el proceso de liofilización también favorece la degradación del ARN. También se evidenció mayor pureza y rendimiento de ARN con el uso de Buffer CTAB y RNeasy mini kit. Por otro lado, los cebadores BtubF y BtubR amplificaron exitosamente regiones codificantes del gen de referencia β-tubulina útiles para llevar a cabo estudios de expresión génica en ascomicetes. Finalmente, se logró obtener ADN complementario (cADN) amplificable mediante RT-PCR independiente de la metodología de homogenización y extracción probada. No obstante, se recomienda la homogenización con nitrógeno líquido y extracción con RNeasy mini kit cuando se busca eficiencia en el manejo de tiempo y calidad del ARN en la evaluación de expresión génica mediante RT-PCR.

