











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
Introducción
¿Todos vamos a desarrollar un cáncer subclínico? Esta era la pregunta que se hacía Mel Greaves y que daba el título a su artículo en la revista Nature Reviews Cancer 1. En este artículo, el autor plantea que alrededor de uno de cada tres individuos, si viven lo suficiente, tendrán un diagnóstico clínico confirmado de cáncer y que existe evidencia que muchos de nosotros desarrollaremos un cáncer subclínico. Lo anterior es de esperar si se tiene en cuenta que la población mundial está envejeciendo. Entre 2015 y 2030, se prevé que el número de personas mayores de 60 años en el mundo aumente en un 56 %, de 901 millones a 1.4 mil millones, y para 2050, se proyecta que la población mundial de personas mayores sea más del doble de su tamaño en 2015, llegando a casi 2,1 mil millones 2. Además, en Estados Unidos, entre 2010 y 2030, se prevé un aumento del 67% en la incidencia de cáncer para pacientes de 65 años o más en comparación con solo un aumento del 11% en la incidencia de cáncer para pacientes menores de 65 años 3.
Conforme las personas envejecen, en los tejidos se acumulan mutaciones somáticas; aunque muchas de estas mutaciones tienen poca o ninguna consecuencia funcional, puede aparecer una mutación que le confiera una ventaja de crecimiento a una célula (mutación conductora) en el microambiente de un órgano de otra manera sano (4, 5). Un clon mutante puede crecer para producir campos de cancerización que como su nombre lo indica tienen predisposición a progresar a un cáncer. Las células que conforman estos campos pueden ser morfológicamente normales o presentar displasia, esto último ocurre con mayor frecuencia en los epitelios tanto escamoso como glandular y se conoce con el nombre de lesión premaligna 5.
Estas mutaciones somáticas podrían ser inducidas por factores ambientales o como el resultado de errores en la replicación del ADN. Tomasetti y colaboradores estudiaron la relación entre el número de divisiones normales de células madre y el riesgo de 17 tipos de cáncer en 69 países de todo el mundo. Sus datos revelaron una fuerte correlación entre la incidencia de cáncer y las divisiones normales de células madre en todos los países, independientemente de su entorno 6.
Esto sugiere que la mayoría de los canceres son el resultado de un proceso estocástico no determinista y es que esta idea, aunque simple no deja de ser interesante. Tiene sentido que en una célula madre que constantemente se está dividiendo exista mayor probabilidad de que ocurran errores en la replicación del ADN y por consiguiente mutaciones. Además, esto tiene una implicación muy importante en el abordaje del cáncer, porque si aceptamos que la mayoría de cánceres ocurren por el azar y no por factores ambientales prevenibles, quiere decir que nuestros esfuerzos no se deben enfocar a la prevención primaria sino a la prevención secundaria que consiste en la detección temprana de estos campos de cancerización.
En la última década, los estudios de secuenciación masiva del ADN han permitido identificar campos de cancerización antes de que ocurran cambios morfológicos. En el epitelio escamoso de la piel normal de pacientes caucásicos expuestos a radiación solar, se han encontrado mutaciones en genes como TP53 7. En un estudio para mejorar el entendimiento de la evolución clonal en Leucemia Mieloide Aguda (LMA) se realizó secuenciación de exoma completo en CMHs de individuos sanos de diferentes edades, sugiriendo que estas células acumulan mutaciones con la edad, muchas de las cuales son compartidas por los pacientes con LMA 8. El proceso de campo de cancerización en CMHs ocurre cuando en un individuo sano una proporción sustancial de sus células sanguíneas derivan de un clon con por lo menos una mutación conductora, lo que se acerca a la definición de HCPI.
El objetivo principal de esta revisión es conocer el estado del arte actual acerca de HCPI en diferentes aspectos, comenzando por precisar los criterios diagnósticos que la definen, su epidemiología, las mutaciones somáticas que con mayor frecuencia se encuentran, los mecanismos de expansión clonal, las implicaciones en el tratamiento de pacientes con cáncer hematológico y no hematológico, y la relación con enfermedades inflamatorias crónicas asociadas con envejecimiento.
Para esta revisión se realizó una búsqueda bibliográfica de artículos en Pubmed, usando las palabras claves descritas previamente. De los resultados obtenidos, se seleccionaron 50 referencias, basados en el factor de impacto de las revistas. El factor de impacto de las referencias citadas varía mucho, entre 1 para Signal Transduct Target Ther. Y 71 para N eng J Med. Se eligieron de la siguiente manera: Todas las publicaciones desde el 2015 aparecidas en revistas de más de 5 de factor de impacto y todas las publicaciones de menos de 5 de factor de impacto que resultaron tener relevancia científica luego de la lectura.
De la Hematopoyesis Clonal (HC) a la hematopoyesis clonal de potencial indeterminado: Definiciones
La hematopoyesis clonal (HC) es cualquier sobrecrecimiento de CMHs. Este proceso no es sinónimo de enfermedad, aunque ocurre en muchas. Así, un paciente con una LMA tiene HC, pero se ha encontrado también en personas asintomáticas 4.
En personas asintomáticas, puede observarse HC por dos mecanismos; reducción del repertorio de células madre, la cual ocurre por la edad, y en la cual hay acumulación de mutaciones aleatorias fuera de los genes conductores conocidos 9.
En el otro tipo de expansión, la HCPI, existe una selección positiva por lo menos por una mutación conductora con riesgo de progresión a malignidad (Figura 1).
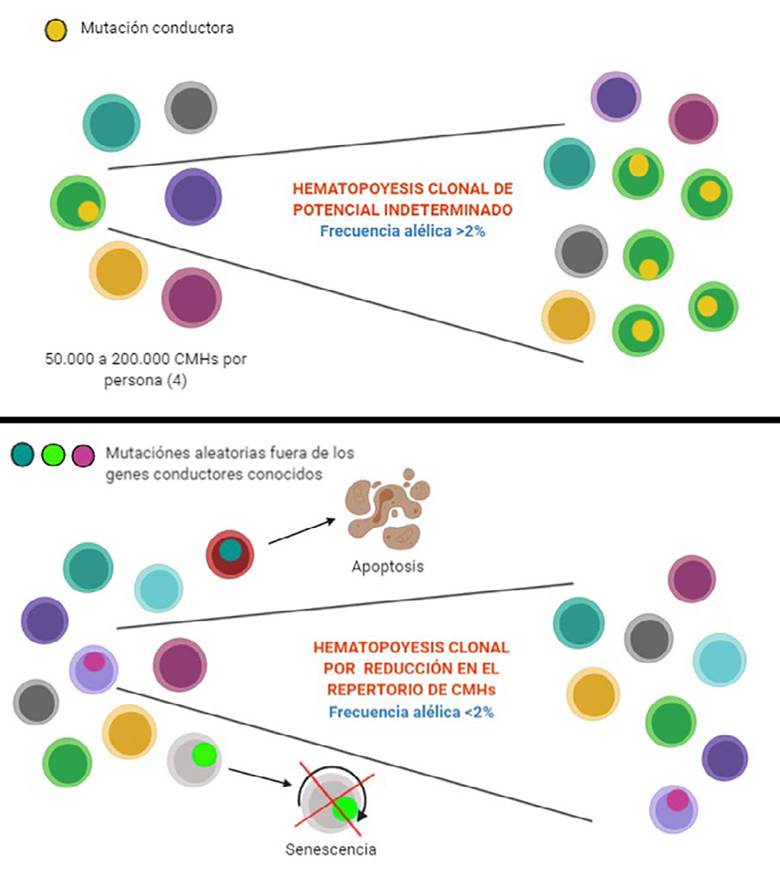
Figura 1 Tipos de hematopoyesis clonal. La hematopoyesis clonal de potencial indeterminado se caracteriza por mutaciones conductoras que llevan a expansión clonal. Por el contrario, en la hematopoyesis clonal por reducción del repertorio de las CMHs, ocurren mutaciones aleatorias fuera de los genes conductores conocidos que llevan a apoptosis o perdida en la capacidad de autorrenovación (senescencia).
Otro aspecto importante en la definición de HCPI es el tamaño del clon mutante; se considera que las consecuencias clínicas de esta entidad son mejor entendidas para clones más grandes con frecuencia alélica mayor a 2% (significa que más del 2% de los alelos secuenciados contienen la mutación o aproximadamente el 4% de las células, si se asume que la mutación es heterocigota) 4. Este umbral tiene que ver con el límite experimental de la secuenciación de nueva generación, la cual no permite identificar de manera confiable mutaciones con frecuencia alélica menores al 2% 10. Debido a que los primeros estudios que encontraron relación de HCPI con progresión a cáncer utilizaron esta tecnología, este umbral ha sido aceptado por algunos autores 11. Otros sugieren que cualquier frecuencia alélica encontrada es válida 12.
Epidemiología de la hematopoyesis clonal
En tres estudios diferentes, realizados en pacientes sin evidencia de cáncer hematológico, se analizaron los datos de secuenciación del exoma completo del DNA de células en sangre periférica con el fin de identificar mutaciones en genes asociados a este cáncer (13 15). Aunque estos estudios presentan diferencias importantes respecto al número y las características clínicas de las personas estudiadas, todos coincidieron en que la frecuencia de las mutaciones asociadas con cáncer hematológico aumenta con la edad, siendo raras en menores de 40 años, pero encontrándose en el 6% al 10% de los mayores de 70 años (Tabla 1).
Tabla 1 Comparación de la prevalencia de hematopoyesis clonal de potencial indeterminado. Se incluyen datos del número de pacientes, características clínicas y relación con algunos desenlaces. N/A no aplica, DM2 diabetes mellitus tipo 2, CR cociente de riesgo, IC intervalo de confianza.
| Xie et al 13 | Jaiswal et al 14 | Genovese et al 15 | |
|---|---|---|---|
| Número de pacientes | 2728 | 17182 | 12380 |
| Plataforma y tipo de muestras utilizada | Secuenciación de exoma completo -DNA en células de sangre periférica | Secuenciación de exoma completo -DNA en células de sangre periférica | Secuenciación de exoma completo -DNA en células de sangre periférica |
| Media y rango de edad en años | 59.5 (10-90) | 58 (19-108) | 55 (19-93) años |
| Características de los pacientes | 2728 pacientes con tumores sólidos, 25% con adenocarcinoma de mama | 15801 pacientes de un estudio de casos y control de DM2, 1381 pacientes del Jack Heart Study | 6245 controles sanos 4970 pacientes con esquizofrenia 1165 pacientes con desorden bipolar |
| Prevalencia de mutaciones según el rango de edad en años | <40: raras 60-69: 2.2% 70-79: 6.1%80-89: 6.8%>70: 5-6% | <40: raras 60-69: 5.6% 70-79: 9.5% 80-89: 11.7%>90: 18.4>70: 10% | <50: 0.9% >65: 10.4% |
| Incremento en el riesgo de cáncer lógico | N/A | CR 11.1; 95% IC, 3.9 a 32.6 | CR 12.9; 95% IC, 5.8 a 28.7 |
| Incremento en el riesgo de evento coronario agudo | N/A | CR 2.0; 95% IC, 1.2 a 3.4 | N/A |
| Incremento en el riego de acódente cerebrovascular isquémico | N/A | CR 2.6; 95% IC, 1.4 a 4.8 | N/A |
| Incremento en la mortalidad por cualquier causa | N/A | CR 1.4; 95% IC, 1.1 a 1.8 | N/A |
En uno de los estudios, hasta el 83% de las mutaciones se encontraron con una frecuencia alélica mayor al 10% 13, lo cual fue consistente con otro donde la mediana de la frecuencia alélica fue 9% 14. Lo anterior demuestra que la mayoría de los clones mutantes tienen un tamaño apreciable. Cuando se utilizan métodos que permiten corregir la tasa de error de la secuenciación de nueva generación, mejorando el umbral de detección de fracción alélica del 2% al 0.03%, la prevalencia de HC aumenta, como lo demostró un estudio en el que de 20 enfermeras entre los 50 y 70 años, el 95% presentaban mutaciones asociadas con cáncer hematológico 16. Sin embargo, el significado clínico de estos clones mutantes pequeños con frecuencias alélicas menores de 2% actualmente no está claramente establecido.
Hematopoyesis clonal como un modelo de mutaciones somáticas asociadas al envejecimiento
La mutación somática que con mayor frecuencia ocurre en los individuos con HC es un cambio de una citosina a timina (C - T) considerada una firma del envejecimiento 14. Esta mutación ocurre por desaminación espontanea que convierte una citosina a uracilo, este último es escindido por la acción de la enzima uracil DNA glicosilasa llevando a una reparación libre de error. Los residuos 5'-metilcitosina son desaminados a timina, la cual no puede ser escindida ni reparada por este sistema 17.
Como se describirá más adelante, tres de los genes que con mayor frecuencia se encuentran mutados en HCPI están implicados en regulación epigenética; por ejemplo, uno de estos genes TET-2 codifica para una enzima desmetilasa de DNA que en condiciones normales lleva a la reparación de la 5-metilcitosina a una citosina no modificada 18. Lo anterior sugiere que en HCPI la epigenética actúa como un mecanismo de retroalimentación positiva, donde la mayoría de las mutaciones ocurren por alteraciones epigéneticas en genes que a su vez están implicados en la regulación de mecanismos epigenéticos.
Mutaciones prevalentes en hematopoyesis clonal
Otro aporte significativo de los estudios iniciales de secuenciación de exoma completo en el escenario de HC consistió en que permitieron identificar los genes que con mayor frecuencia se encuentran mutados (Tabla 2). El top cinco en orden descendente de frecuencia lo conforman:
Tabla 2 Genes más frecuentemente mutados en hematopoyesis clonal de potencial indeterminado.
| Xie et al 13 | Jaiswal et al 14 | Genovese et al 15 | |
|---|---|---|---|
| Número total de mutaciones encontradas | 78 | 805 | 3111 |
| Genes más frecuentemente mutados (número de mutacionesencontradas) | DNMT3A (18) | DNMT3A (403) | DNMT3A (190) |
| TET2 (10) | TET2 (72) | ASXL1 (35) | |
| JAK2 (8) | ASXL1 (62) | TET2 (31) | |
| ASXL1 (6) | TP53 (33) | JAK2 (24) | |
| TP53 (4) | JAK2 (31) | PPM1D (15) | |
| GNAS (3) | SF3B1 (27) | SF3B1 (13) | |
| PPM1D (2) | GNB1 (22) | SRSF2 (7) | |
| BCORL1 (2) | CBL (12) | TP53 (4) | |
| SF3B1 (2) | SRSF2 (11) | CBL (3) | |
| GNAS (8) | MYD88 (1) | ||
| U2AF1 (1) | |||
| STAT3 (1) | |||
| IDH2 (1) | |||
| ATM (1) |
1. DNMT3A (que codifica la enzima ADN (citosina-5)-metiltransferasa 3A).
2. TET-2 (que codifica la enzima tet metilcitosina dioxigenasa 2).
3. ASLX-1 (que codifica el regulador transcripcional adicional combs-like 1).
4. JAK2 (que codifica la proteína Janus quinasa 2).
5. TP53 (que codifica la proteína tumoral p53) (13-15).
Es de anotar que mutaciones en genes importantes como JAK2 y TP53 sean menos frecuentes que las encontradas en otros genes, teniendo en cuenta que las mutaciones con cambios de sentido en JAK2 son las más prevalentes en pacientes con Neoplasias Mieloprolferativas Crónicas (NMPCs) philadelphia negativo (BCR-ABL negativo) 19 y que mutaciones somáticas en TP53 están presentes en el 10% de los casos de síndromes mielodisplásicos (SMDs) y LMA y 30% de los pacientes con Neoplasia Mieloide Asociada a Terapia (NMAT) 20.
La presencia de estas mutaciones conductoras es relevante para iniciar la expansión clonal de HCPI y corresponden a eventos de inicio temprano para cánceres hematológicos como NMPCs, SMDs y LMA. Otras mutaciones asociadas a NMPCs, SMDs y LMA (NRAS, RUNX1, NPM1 y FLT3) son mutaciones cooperativas que están involucradas en la progresión de estas neoplasias 8,15.
¿Cómo las mutaciones en estos genes llevan a expansión clonal? Este sigue siendo en la actualidad un tema de constante debate.
Mecanismos de hematopoyesis clonal
Las CMHs pueden autorrenovarse y diferenciarse al mismo tiempo vía división asimétrica; esta vía limita su capacidad de expansión durante el desarrollo embrionario. Por otro lado, las CMHs pueden dividirse vía simétrica dando origen a dos células indiferenciadas que continúan siendo células madre; esta vía es la que predomina después de un trasplante de médula ósea. Sin embargo, en condiciones normales, en la médula ósea ocurren más divisiones asimétricas, manteniendo un número más o menos constante de CMHs (Figura 2) 21.

Realizada por: Oscar Andrés Franco Javera
Figura 2 Vías de división en Células Madre Hematopoyéticas (CMHs). La vía de división simétrica da origen a dos células indiferenciadas promoviendo la expansión de las CMHs, mientras que la vía de división asimétrica da origen a una célula indiferenciada y otra diferenciada evitando la expansión de las CMHs.
Las mutaciones en TP53 descritas en HCPI confieren ventajas competitivas a las CMHs trasplantadas a ratones receptores letalmente irradiados, favoreciendo la división simétrica. El mecanismo propuesto implica una vía epigenética, y es que p53 mutado interactúa con la histona metiltransferasa EZH2 favoreciendo su asociación con la cromatina, aumentando así los niveles de H3K27me3 en genes que regulan la autorrenovación y diferenciación de las CMHs 20. Además, este mecanismo podría explicar los casos de NMAT en pacientes con HCPI y mutaciones en TP53 tratados con trasplante de médula ósea autólogo para linfomas 22.
Las mutaciones en el gen PPM1D, un regulador de las respuestas de daño al ADN, confiere ventajas de supervivencia en CMHs en el contexto de pacientes con NMAT expuestos a cisplatino, mediado por incremento de la resistencia a la apoptosis. Se desconocen los mecanismos 23.
La presencia de la mutación JAK2 V617F encontrada en NMPCs Ph- confiere una ventaja proliferativa y de supervivencia al hacer que las CMHs sean más sensibles a las señales estimuladoras de esta vía, como la iniciada por la interleucina-3 (IL-3) 24.
Aunque en las células normales los genes DNMT3 y TET2 codifican para enzimas que cumplen funciones diferentes con actividad ADN metiltransferasa y ADN desmetilasa, respectivamente, mutaciones con pérdida de la función en estos dos genes activan genes asociados a autorrenovación y silencian otros asociados a diferenciación en CMHs 25, 26.
Se desconocen los mecanismos epigenéticos precisos por los cuales la metilación o desmetilación de las regiones promotoras llevan a la activación de los genes asociados con autorrenovación.
Hematopoyesis clonal y transformación a cáncer hematológico
Los SMDs son trastornos clínicamente heterogéneos caracterizados por HC, displasia de células de la médula ósea y sangre periférica, citopenias y un riesgo de progresión a LMA 27. La HCPI y los SMDs no solo comparten su naturaleza clonal sino también muchas mutaciones.
Mutaciones asociadas a HCPI como TET2 y ASXL1 son frecuentemente encontradas en pacientes con SMD y LMA con cambios relacionados con displasia (27-29). Por el contrario, en pacientes con LMA de novo, solo DNMT3A se encuentra mutado con frecuencia 30 (Tabla 3). Así, las mutaciones en genes como TET2 y no solo en la progresión a SMD sino también a LMA ASXL-1, en individuos con HCPI, estarían implicadas como un evento tardío.
Tabla 3 Comparación de las mutaciones más frecuentes en Hematopoyesis Clonal de Potencial Indeterminado con las encontradas en varios cánceres hematológicos. Las mutaciones están en orden descendente de frecuencia.
| Hematopoyesis Clonal de Potencial Indeterminado (HCPI) (13-15) | Síndromes Mielodisplásicos (SMDs). Bejar et al 27 | Síndromes Mielodisplásicos (SMDs). Papaemmanuil et al 28 | Leucemia Mielode Aguda (LMA) con cambios asociados a displasia. 29 | Leucemia Mieloide Aguda (LMA) de novo. 30 |
| DNMT3A | TET2 | SF3B1 | ASXL1 | FLT3 |
| TET2 | ASL1 | TET2 | RUN1 | NPM1 |
| ASL1 | RUN1 | SRS2 | TET2 | DNMT3A |
| A2 | TP53 | ASL1 | IDH1 | IDH2 |
| TP53 | EZH2 | DNMT3A | IDH2 | IDH1 |
Los estudios de Genovese y Jaiswal han demostrado que la presencia de mutaciones en el contexto de HCPI se asocia con aumento en el cociente de riesgo de desarrollar cáncer hematológico entre 12.9 y 11.1, respectivamente, comparado con personas de la misma edad sin mutaciones, con una tasa de transformación anual de 0.5-1% 14,15. Esta tasa de transformación es similar a la reportada en otros estados clonales conocidos por ser precursores de cáncer hematológico, como gammapatía monoclonal de significado incierto, que es precursor de mieloma múltiple y linfocitosis monoclonal de células B, que es precursor de la leucemia linfocítica crónica y otros linfomas B 31.
Existen por lo menos tres variables importantes que influyen en el riesgo de transformación de malignidad en individuos con HCPI: 1. Genes mutados: mutaciones en TP53 y U2AF1 se asocian con un aumento en el riesgo relativo de desarrollar LMA en 47.2 y 7.4, respectivamente 32. 2. Tamaño del clon mutante: varios estudios han demostrado consistentemente que mutaciones somáticas en genes asociados a HCPI con una frecuencia alélica mayor al 10% tienen mayor riesgo de transformación maligna 14,32-33. 3. Número de mutaciones: Albeson utilizó un modelo para predecir la sobrevivida libre de LMA, encontrando que los individuos con HCPI y más de un gen mutado tenían un riesgo relativo aproximado de 2.5 de desarrollar LMA comparado con los que tenían una sola mutación 33.
En el estudio de Xie y colaboradores, la búsqueda de mutaciones asociadas a HCPI se realizó en pacientes con diagnóstico de 11 tipos diferentes de cáncer, incluyendo carcinomas y gliomas de bajo y alto grado. Aunque sus resultados evidenciaron que el 5-6% de las personas mayores de 70 años tenían mutaciones relacionadas con HC, no se investigó la asociación con las terapias recibidas ni el impacto de estas mutaciones en los resultados clínicos de los pacientes 13. En 2017, Coombs y colaboradores realizaron secuenciación de nueva generación en muestras de sangre y del tumor en 8.810 pacientes con cáncer no hematológico con resultados similares a los encontrados por el estudio de Xie y colaboradores. Sin embargo, ellos profundizaron un poco más demostrando que las mutaciones en los genes PPM1D y TP53 tenían una asociación clínicamente significativa con exposición a quimioterapia y radioterapia 34.
El papel de TP53 en el origen y la evolución de NMAT ha sido establecido en varios estudios (20, 35). Por su parte mutaciones en el gen PPM1D fueron encontradas en el 20% de los casos de NMAT, siendo la segunda mutación más prevalente después de TP53 23. El riesgo de NMAT no solo se encuentra en pacientes con tumores sólidos o cáncer no hematológico tratados con quimioterapia y radioterapia, sino en pacientes con trasplante de médula ósea autólogo como parte del tratamiento de linfomas. Esto último se demostró en un estudio de 12 pacientes en el cual la presencia de mutaciones en PPM1D y TP53 fueron más frecuentes si se comparan con otros estudios que incluyen individuos con HCPI en la población general 22. Esto probablemente se debe a selección clonal después que estos pacientes fueron sometidos a quimioterapia y sus CMHs recolectadas para el trasplante autólogo.
El cáncer no hematológico más común asociado con HC es el carcinoma de tiroides, con una prevalencia de 36% 34 y en un estudio, la frecuencia de HCPI en pacientes con carcinoma de tiroides aumentó con relación a la edad y a la dosis de yodo radioactivo recibida. Además, se asoció a disminución en la supervivencia 36. La presencia de HCPI con mutaciones en los genes TP53 y PPM1D se puede considerar entonces como un factor predictivo para la ocurrencia de NMAT y un factor de pobre pronóstico, tanto en cáncer hematológico como no hematológico, tratadas con quimioterapia, radioterapia o trasplante de médula ósea.
Hematopoyesis clonal y enfermedad cardiovascular ateroesclerótica
La HC se asocia con aumento en la mortalidad por cualquier causa (cociente de riesgo, 1.4) e incrementa el riesgo de síndrome coronario (cociente de riesgo, 2.0) y accidente cerebrovascular isquémico (cociente de riesgo, 2.6) 14. Otros estudios también han encontrado aumento en la mortalidad en individuos con HC por diversas causas; en los pacientes que no desarrollaron cáncer hematológico las muertes estaban relacionadas con el hábito de fumar 15,37.
Esto planteó dos posibilidades: 1. Que HCPI y las enfermedades cardiovasculares (ECV) compartieran factores de riesgo como el hábito de fumar o 2. que la HCPI estuviera implicada en la fisiopatología de las ECV. Dado que la ateroesclerosis es una enfermedad inflamatoria crónica que se caracteriza por la acumulación de macrófagos cargados con lípidos en la íntima de las arterias, los cuales derivan de los monocitos de la circulación 38, esto implicaría que los clones mutados dan origen a células sanguíneas incluyendo células inflamatorias que pueden interactuar con diferentes tejidos 4.
Utilizando un modelo murino de ateroesclerosis se evidenció que la pérdida de función de TET2 en las CMHs acelera el proceso de aterogénesis, al parecer a través del mejoramiento en el reclutamiento de monocitos y otras células sanguíneas a sitios periféricos, incluyendo la íntima arterial, debido al aumento en la expresión de quimiocinas por parte de los macrófagos tisulares con mutaciones en TET2 39.
Hematopoyesis clonal en otras condiciones
Cook y colaboradores, en un estudio de casos y controles, encontraron que individuos con HCPI que presentaban más de una mutación, incluyendo una mutación en DNMT3A, tenían un número mayor de comorbilidades que el grupo control. Además, los niveles séricos de interleucina 6 (IL-6) en los individuos con HCPI que presentaban mutaciones en TET2 fueron dos veces más altos que los del grupo que no presentaba mutaciones 40. Entre las dolencias encontradas estaban la enfermedad pulmonar crónica y diabetes con compromiso de órgano blanco; entidades que previamente se habían asociado a HCPI 14,15,37,39,41 y reflujo gastroesofágico sumándose por primera vez a la lista.
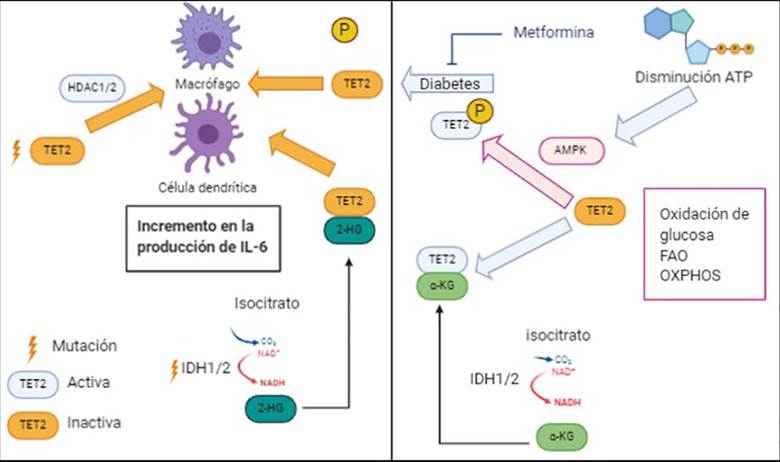
Los ratones deficientes de TET2 son más susceptibles a la colitis inducida por dextrano sulfato sódico y endotoxinas con un incremento en la producción de IL-6 en células dendríticas y macrófagos, comparados con ratones que presentan TET2 silvestre 42. TET2 se ha convertido en un modelo de como las vías inflamatorias, metabólicas y epigenéticas interactúan en cáncer (Figura 3). AMPK (proteína quinasa activada por AMP) es un sensor de energía y nutrientes activado en respuesta a la depleción de energía para restaurar los niveles de ATP en la célula, pasando de un metabolismo anabólico a catabólico 43. La proteína TET2 es fosforilada por AMPK en la serina 99, produciendo su estabilización que es indispensable para su función de metilcitosina dioxigenasa.
En los pacientes diabéticos, el aumento en los niveles de glucosa impide la fosforilación de TET2 en la serina 99 por AMPK con la consiguiente desregulación; sin embargo, medicamentos utilizados en diabetes como la metformina favorecen la fosforilación de TET2 incrementando su estabilidad y manteniendo su función 44.
La función de la proteína TET2 también es regulada por alfa-cetoglutarato, que actúa como un cofactor. El alfa-cetoglutarato es un intermediario del ciclo de Krebs producido a partir del citrato por acción de la isocitrato deshidrogenasa (IDH) que presenta dos isoformas IDH1/2 21. Las mutaciones en los genes
Realizada por: Oscar Andres Franco Tavera
Figura 3 Pérdida de función de TET2. Existen varios mecanismos que pueden terminar en pérdida de función de TET2: 1. Mutaciones en el gen TET2 que impiden el reclutamiento de las Histonas Deacetilasas 1 y 2 (HDAC1/2). 2. La falta de fosforilación de la proteína TET2 por la proteína quinasa activada por AMP (AMPK) como ocurre en pacientes con diabetes, y 3. Por la unión de la proteína TET2 con el oncometabólito 2-hidroxiglutarato (2-HG) producido por mutaciones en los genes que codifican las enzimas isocitrato deshidrogenases 1 y 2 (IDH1/2). Independientemente del mecanismo de perdida de función de TET2, esta lleva al incremento en la producción de interleuquina 6 (IL-6) en macrófagos y células dendríticas.
IDH1/2 frecuentes en glioblastomas y LMA llevan a la acumulación del oncometabólito 2-hidroxiglutarato, el cual es un inhibidor competitivo de dioxigenasas dependientes de alfa-cetoglutarato como TET2 45.
El envejecimiento humano se caracteriza por una inflamación crónica de bajo grado, fenómeno denominado inflammaging, lo cual es respaldado por la evidencia epidemiológica que demuestra que el aumento en biomarcadores inflamatorios como la proteína C reactiva e IL-6 es asociado y predictivo de envejecimiento 46. La HCPI podría por lo menos en parte explicar este estado proinflamatorio que se da durante el envejecimiento porque, como vimos antes, las mutaciones en TET2 favorecen la liberación de IL-6 y al mismo tiempo fuentes como infecciones o comorbilidades preexistentes pueden favorecer la expansión clonal 47.
Las enfermedades autoinmunes también se han asociado a HCPI. La Anemia Aplásica (AA) adquirida es causada por destrucción de CMHs y células progenitoras a través de mecanismos inmunes. En la medida en que los pacientes con AA han mejorado la supervivencia, hasta un 15% de ellos pueden desarrollar SMDs o LMA; un fenómeno llamado evolución clonal. Como era de esperarse en un estudio que incluyo 439 pacientes con AA de diferentes países, se encontró que la prevalencia de mutaciones somáticas fue del 36% con un tercio de estos pacientes teniendo más de una mutación y siendo los tres genes más mutados: BCOR, BCORL1 y PIGA 48. Llama la atención que el tamaño de estos clones mutantes fue menor a una frecuencia alélica del 10%, permaneciendo así a través del tiempo o incluso desapareciendo, y se asociaron con una mejor respuesta a la terapia inmunosupresora. En este caso, las mutaciones de estos genes, más que conferir a las CMHs una ventaja de sobrecimiento parece hacerlas más resistentes a la destrucción por los linfocitos T, similarmente a lo que ocurre en pacientes con hemoglobinuria paroxística nocturna, un tipo de HC caracterizado por mutaciones somáticas en el gen PIGA que lleva a deficiencia de glicosilfosfatidilinositol 49.
En un estudio de 59 pacientes con artritis reumatoide (AR), la prevalencia de HCPI fue del 17% con mutaciones en DMNT3A y TET2 siendo las más frecuentes; sin embargo, no se encontraron diferencias con respecto a los tratamientos recibidos y los desenlaces comparados con los pacientes con AR que no presentaban HCPI 50.
Conclusiones
La HCPI es un modelo de campo de cancerización que ha atraído la atención de los científicos en la última década. En parte, este interés deriva de la idea que estos clones mutados dan origen a diferentes tipos de células sanguíneas con funciones alteradas que predisponen al desarrollo de las dos principales causas de mortalidad en el mundo: el cáncer y las enfermedades cardiovasculares. Ambas se consideran enfermedades crónicas, aumentan con la edad y están dentro de la cuenta de alto costo. Es así como la mayoría de las mutaciones somáticas en HCPI ocurren como una firma del envejecimiento y que los tres genes mutados con mayor frecuencia participan en la regulación epigenética.
Como hemos visto en esta revisión, se ha avanzado en el entendimiento de los mecanismos de expansión clonal a través de los procesos de envejecimiento, inmunidad y de las terapias citotóxicas. Sin embargo, poco se conoce de los factores ambientales relacionados con la HCPI, incluyendo el impacto del microambiente de la médula ósea y tampoco se ha explorado la asociación con enfermedades neurodegenerativas como Alzheimer y Parkinson.
Otra oportunidad es el papel que la HCPI puede jugar como factor predictivo de desenlace en pacientes con cáncer hematológico y no hematológico que son tratados con quimioterapia y/o radioterapia dada su asociación con NMAT. En un futuro, se podrán desarrollar terapias dirigidas a esta población vulnerable con el fin de detener la progresión de malignidad, con los impactos que esto tendría disminuyendo los costos en salud pública destinados a los pacientes con un cáncer plenamente establecido.
Por último, aunque algunos estudios sugieren una prevalencia menor de HCPI en poblaciones hispanas 14, no hay estudios en Latinoamérica que así lo confirmen ni tampoco conocemos cuales son las mutaciones más prevalentes entre nosotros

