










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActualidades Biológicas
Print version ISSN 0304-3584
Actu Biol vol.33 no.94 Medellín Jan./June 2011
ARTÍCULOS DE INVESTIGACIÓN
MODELACIÓN ESTRUCTURAL DE LA PROTEÍNA
DE LA CÁPSIDE DEL VIRUS A DE LA PAPA (PVA, POTYVIRUS)
STRUCTURAL MODELLING OF POTATO VIRUS A (PVA, POTYVIRUS) COAT PROTEIN
Pablo Gutiérrez1; Sara Bastos-Aristizábal2; Mauricio Marín3
1 Laboratorio de Microbiología Industrial, Facultad de Ciencias. Universidad Nacional de Colombia (Sede Medellín). Medellín (Antioquia), Colombia. paguties@unal.edu.co.
2Laboratorio de Microbiología Industrial, Facultad de Ciencias. Universidad Nacional de Colombia (Sede Medellín). Medellín (Antioquia), Colombia. sarabastos111@gmail.com.
3 Laboratorio de Biología Celular y Molecular, Facultad de Ciencias. Universidad Nacional de Colombia (Sede Medellín). Medellín (Antioquia), Colombia. mamarinm@unal.edu.co.
Recibido: agosto 2010; aceptado: mayo 2011.
Resumen
A diferencia de lo que ocurre con diversos virus icosahédricos, la estructura a alta resolución de la cápside de los virus flexuosos de plantas pertenecientes a la familia Potyviviridae no ha podido ser determinada aún. Los potyvirus son un grupo de gran importancia económica en la agricultura al afectar cultivos como papa, tomate, tabaco, papaya y caña de azúcar, entre muchos otros; por lo cual la comprensión de su estructura puede arrojar información valiosa para lograr un conocimiento más detallado de sus mecanismos biológicos, con miras al diseño de estrategias de control. En este trabajo se presenta un modelo de la estructura tridimensional de la región central de la proteína de la cápside del virus A de la papa (PVA), utilizando una combinación de herramientas de predicción de estructura secundaria y docking. El modelo presentado tiene dimensiones compatibles con la estructura de baja resolución obtenida en otros estudios mediante microscopía electrónica y será de gran utilidad en el diseño de experimentos de mutagénesis dirigida, enfocados en el estudio del ensamblaje de la partícula viral y como base para modelar la estructura de otras especies potyvirales de importancia actual en Colombia como el virus Y de la papa (PVY), virus de la malformación de las hojas del tomate de árbol (TaLMV) y el virus de la mancha anular de la papaya (PRSV).
Palabras clave: cápside viral, modelo estructural, potyvirus, PVA
Abstract
In contrast to icosahedric viruses, a high-resolution structure of flexuous viruses of plants belonging to the family Potyviridae has yet to be obtained. Potyviruses are economically important in agriculture due to their ability to infect crops such as potato, tomato, tobacco, papaya, and sugar cane, among many others; thus an understanding of its three-dimensional structure might provide valuable insight into biological mechanisms, with the aim of designing control strategies. In this study, we present a model for the three-dimensional structure of the core region of Potato virus A (PVA), using a combination tool to predict the secondary structure and docking. The model presented has dimensions compatible with the low resolution structure in other studies using electron microscopy and will be highly useful in the design of experiments of directed mutagenesis aimed at understanding potyvirus assembly and as a basis for modelling the structure of other important related virus species of importance in Colombia, such as Potato virus Y (PVY), Tamarillo leaf malformation virus (TaLMV), and Papaya ringspot virus (PRSV).
Key words: virus capsid, structural model, Potyvirus, PVA
INTRODUCCIÓN
El genero Potyvirus de la familia Potyviridae es uno de los grupos de virus mas numerosos y limitantes de diversos cultivos agricolas. Los potyvirus se caracterizan por tener estructura de varillas flexuosas de 650 a 900 nm de longitud y 11 a 13 nm de ancho. Su genoma es de ARN de cadena sencilla positiva con tamano aproximado de 9-10 kpb, poliadenilados en su extremo 3' y asociados a una proteina unida covalentemente al extremo 5' (VPg) (Singh et al. 2008, Van- Regenmortel et al. 2000). El genoma de los miembros del genero Potyvirus codifica para una poliproteina precursora de 350 kDa (Riechmann et al. 1995), que es subsecuentemente procesada en siete proteinas pequenas: P1, componente asistente (Helper Component); P3, de inclusion cilindrica (CI); inclusion nuclear A (NIa); inclusion nuclear B (NIb); proteina de capside (CP); y dos proteinas putativas conocidas como 6K1 y 6K2. El clivaje es llevado a cabo por acción de tres proteasas de origen viral: la proteinasa P1 y la proteinasa del componente asistente (HCpro), catalizan solo reacciones autoproteoliticas en los extremos C terminales (Carrington et al. 1989, Urcuqui-Inchima et al. 2001, Verchot et al. 1991) y los clivajes restantes son catalizados mediante mecanismos trans y autoproteoliticos mediados por la proteina de inclusion nuclear (NIa-Pro), un homologo de la proteinasa del picornavirus 3C (Carrington y Dougherty 1987, Langenberg y Zhang 1997). El procesamiento y función de todas estas proteinas es aun controvertido pero se cree que muchas de ellas son multifuncionales (Riechmann et al. 1992, Verchot y Carrington 1995).
La principal función de la proteina CP es encapsidar el ARN viral y evitar que sea degradado. Se cree que la interacción específica entre el ARN viral y CP es el principal factor influyente en la encapsidación selectiva del genoma viral en medio de gran cantidad de ARN celular (Duggal y Hall 1993, Lin et al. 2009). Sin embargo, Merits (1998), reporto que varias proteinas virales; entre ellas CP, tambien pueden unirse de manera inespecifica al ARN. La CP de los potyvirus se ha dividido en tres dominios principales: el N-terminal variable y C-terminal, expuestos al exterior del virion y sensibles a tratamientos con tripsina y el dominio globular central, el cual es el mas conservado. El dominio N-terminal, aunque no esta involucrado directamente en la arquitectura del virion, es necesario para la transmision de los potyvirus por insectos vectores de la familia Aphididae y ademas es el dominio que contiene mas epitopes virus-específicos (Lin et al. 2009, Mink et al. 1999, Rajamaki et al. 1998).
Pese a que en las últimas decadas ha habido gran progreso en la determinación estructural de virus icosahedricos, el estudio de los virus filamentosos, con la excepción de los tobamovirus y los bacteriofagos filamentosos, ha tenido poco progreso (Ehrenfeld et al. 2008). Es asi como no existe aun un modelo tridimensional que permita explicar los patrones de conservación de la secuencia, ni la manera como las unidades se asocian para formar la estructura viral. Esta información es fundamental para el soporte de estudios sobre la biologia del virus y su interacción con hospedantes y vectores biologicos. En este trabajo, se presenta el modelo de la estructura tridimensional de la region central de la proteina de la capside del PVA (virus A de la papa), un potyvirus que causa mosaicos suaves en diferentes variedades de papa (Solanum tuberosum), y para el cual se han propuesto algunos modelos que contrastados con estudios recientes de microscopia electronica, no concuerdan con las dimensiones esperadas para los componentes de la capside viral (Kendall et al. 2008). Este modelo servira de base para la construcción de estructuras por homologia de otros potyvirus de importancia economica en Colombia, especialmente de aquellas especies virales que afectan los cultivos de solanaceas como la papa, el tabaco y el tomate de arbol.
MATERIALES Y METODOS
Alineamientos de secuencia y predicción de la estructura secundaria. El alineamiento de secuencias se realizo con el modulo CLUSTAL del programa BIOEDIT (Hall 1999) utilizando la matriz de sustitución BLOSUM62 (Henikoff y Henikoff 1992). Se utilizaron las siguientes secuencias de potyvirus, obtenidas de Gen- Bank: virus de la mancha anular de la papaya (PRV), gi: 9629244, virus Plum pox (PPV), gi: 9626508, virus Y de la papa (PVY), gi: 9627728, virus del mosaico del nabo (TuMV), gi: 56407093, virus del mosaico de la soya (SMV), gi:12018225, virus del grabado del tabaco (TEV), gi: 9790340. Para la predicción de estructura secundaria se utilizaron tres metodos diferentes: GORIV (Doolittle et al. 1996), HNN y PredictP (Rost et al. 2004). La predicción de regiones desordenadas se hizo con el programa DisEMBL (Linding et al. 2003a, b) con una ventana de ocho residuos para el algoritmo Savitzky-Golay y el valor umbral de 1,20. La entropia del alineamiento multiple fue medida con la ecuación de Shannon y se grafico como un valor de Z utilizando un promedio local de 5 aminoacidos. La hidrofilicidad fue estimada mediante un grafico de los valores de Hopp y Woods para cada aminoacido con un promedio local de 9 aminoacidos (Hopp y Woods 1981). La confiabilidad de las predicciónes fue verificada utilizando la estructura del virus del mosaico del tabaco (TMV) como control (PDBid: 2TMV) (Namba y Stubbs 1986).
Modelamiento estructural. La estructura del nucleo central de la proteina de la capside del potyvirus fue modelada utilizando una estrategia de docking secuencial, que consistio en el empaquetamiento gradual de cada una de las helices utilizando una rutina de docking de cuerpo rigido con el programa Autodock 4,0 (Morris et al. 1998). Para el docking se utilizo un algoritmo genetico lamarquiano y se realizaron 100 simulaciones con una población inicial de 150 moleculas y un maximo de 2.500.000 evaluaciones de energia. Las tasas de mutación y entrecruzamiento fueron de 0,02 y 0,8, respectivamente. A cada modelo obtenido durante cada paso de docking se le adicionaron las regiones conectoras con el programa MODELLER 9v2 (Sali y Blundell 1995) y se realizo minimización de energia con el programa NAMD utilizando el campo de fuerzas CHARMM27 (Phillips et al. 2005). El modelo final fue minimizado mediante un calentamiento gradual de 0 a 310 K en condiciones periodicas de solvente. La calidad del modelo fue evaluada con el programa WHAT IF (Hooft et al. 1996). Todas las ilustraciones estructurales fueron generadas con PyMOL (DeLano 2002).
RESULTADOS
Validación de las herramientas de predicción de estructura secundaria. Antes de llevar a cabo la predicción de estructura secundaria para la proteina de la capside de potyvirus, se verifico su precision y confiabilidad utilizando el virus del TMV como control (datos no mostrados). Varios estudios han establecido que la arquitectura de la capside de los miembros de la familia Potyviridae es muy similar a la de los virus de varilla rigida como el TMV, del cual se encuentra disponible la estructura cristalografica (Nemykh et al. 2008, Shukla y Ward 1989). La parte central de la capside del TMV esta compuesta por un agregado de cuatro helices, LS, RS, RR y LR, que comprende los residuos 19-32, 38-48, 111-135 y 73-87, respectivamente. Tambien existen otros pequenos segmentos helicoidales y una lamina beta ubicados en los extremos N y C terminal. TMV posee un asa interna de estructura desordenada en la forma monomerica que comprende los residuos 97- 113 y esta involucrada en la union a los grupos fosfato del ARN.
Las predicciones de estructura secundaria de los programas GOR, HNN y PHD fueron contrastadas con la estructura real del TMV. Los tres programas funcionaron bien en la predicción de las helices del dominio central mas no en su extension exacta, lo que es de esperar ya que los metodos para la predicción de estructura secundaria tienen precision entre el 60 y el 80% (Rost 2001). Se encontro que al establecer un consenso entre las tres herramientas de predicción se puede estimar la posición de las helices centrales con un margen de error de ± 3 residuos. Sin embargo, los tres programas fallaron en la predicción de las helices externas, lo cual es un indicativo de que su plegamiento depende de interacciones de tipo terciario que no pueden ser predichas por este tipo de programas. De la misma manera, la predicción de estructuras beta laminares tuvo buena correspondencia con las estructuras extendidas segun el grafico de Ramachandran.
Predicción de estructura secundaria. Las proteinas de la capside de los potyvirus tiene un tamano que varia entre 260 y 330 aminoacidos, con un porcentaje de identidad de aproximadamente 75% (figura 1). La region de mayor variabilidad se localiza en el extremo N-terminal que es el elemento determinante de la transmision por vectores de los potyvirus y el analisis de la estructura secundaria indica que esta region solo tiene tendencia a la formación de una pequena helice alfa en el segmento 7-13, pero su baja conservación sugiere que esta no es una caracteristica global de todas las estructuras de capside (figura 2). Igualmente, varios estudios han demostrado que el dominio N-terminal no esta involucrado en la integridad estructural de potyvirus, ni participa de manera significativa en el ensamblaje de la particula viral (Dolja et al. 1994). Por todo lo anterior, es justificable no incluir este dominio en el modelamiento estructural.
Para el dominio central encontramos cinco regiones con tendencia a la formación de helices (α1a, 85-102; α1b, 105-114; α2, 157-177; α3, 190-204; y α4, 213-228). La α1a esta compuesta por seis aminoacidos completamente conservados (S83, N84, R86, F92, W95 y V99) y se observa conservación de la carga en la posición 97, donde solo se encuentran aspartatos y glutamatos. La α1b se caracteriza por la presencia de alto contenido de residuos acidicos y solamente un residuo, M110; esta conservado completamente en las secuencias comparadas. La predicción de helices alfa para otras secuencias de la capside y su bajo nivel de conservación, indican que la helice α1b no siempre esta presente (datos no mostrados); sin embargo, basados en los contornos de microscopia electronica de Kendall et al. (2008), es una hipotesis razonable asumir que α1b es simplemente una prolongación de α1a. Algo similar ocurrio con la predicción de la helice larga de TMV que los programas de estructura secundaria tienden a dividirse en dos helices. Por esta razon se ha modelado α1a y α1b como una sola helice, α1. Las helices α2, α3 y α4 comprenden algunas de las regiones mas conservadas de los potyvirus y no hay duda de que alli se agrupan la mayoria de las interacciones responsables de la estabilidad estructural. El dominio central presenta tres regiones de estructura irregular, que a diferencia de la region N-terminal, son muy conservadas. Las regiones conectoras de las helices α2-α3 y α4-α5 estarian ubicadas en el interior del virus.
El extremo C-terminal es altamente conservado y presenta poca variabilidad en longitud. La predicción de estructura secundaria sugiere la posible presencia de dos laminas beta o estructuras extendidas, β3 y β4, en las posiciones 234-238, 240-243; y una helice terminal, α6, que comprende los residuos 255-264. El analisis de hidrofilicidad sugiere que β4 es altamente polar y probablemente expuesta; lo cual tiene sentido dado su bajo nivel de conservación. El analisis de regiones no globulares sugiere la presencia de un elemento irregular entre las helices α5 y α6. La función de la region C-terminal no es del todo clara, pero se ha demostrado que mutaciones y deleciones en esta region del Virus del mosaico de la soya inhibe la interacción entre subunidades de la capside (Kang et al. 2006).
Modelo de la proteina de la capside de PVA. Debido a la ausencia de estructuras de alta resolución en el Protein Data Bank con al menos el 25% de identidad con la secuencia de potyvirus, no fue posible llevar a cabo la modelación por homologia de esta proteina. Por esta razon, se implemento un procedimiento que consistio en la obtención de un consenso de estructura secundaria y su posterior ensamblaje terciario mediante un procedimiento iterativo de docking y minimización de energia, tal como se describe en la figura 3. En el ultimo paso se adicionaron las regiones no estructuradas y se minimizo el modelo. La calidad de la estructura fue evaluada con el programa WHAT_CHECK (Hooft et al. 1996). Los valores de Z para el grafico de Ramachandran y la conformación del esqueleto de la proteina (2,074 y -0,164, respectivamente), estan en el rango aceptable. El valor de Z para el empaquetamiento de aminoacidos es de -2,148, que esta dentro del rango aceptado para modelos construidos por homologia (-2,0 y -3,0).

El modelo de la region central de la capside del virus PVA se ilustra en la figura 4. El nucleo central esta compuesto por las helices α1, α2, α3 y α4. La interacción entre estas helices estaria mediada por interacciones de tipo hidrofobico donde F92 (α1) se intercala en la hendidura formada por L168 y Y172 de la helice 2, W95 interactua con H164, A167 y L168, M100 con I213 y Y103 con I161. Es interesante resaltar que la helice 1 presenta en sus extremos N y C terminales unos parches de carga positiva y negativa compuestos por los residuos R86, H89, E104, E106, E107, E111 y que el mismo patron es observado para la helice α2, donde el parche basico esta conformado por H153, K155 y R159, mientras que el negativo por E170 y E174. Esta distribución de cargas en ambas helices sugiere una posible interacción intermolecular entre subunidades de la capside por complementariedad de cargas y potencialmente importante en el ensamblaje de potyvirus (figura 4). La helice 2 interactuaria con la helice 3 a traves de los residuos L188, R193, S196, L197, Y200 y F204. F165 se intercala entre los residuos Y200 y F204; A169 interactua con I197, I173-L188, mientras que R193 formaria un par ionico con E170. Las interacciones de α3 con α4 se podrian dar a traves de los residuos H219, L220, K223, A224 y L227. El residuo D203 y E206 forma interacciones de carga complementarias con H219, K223 y R199 (α3), mientras que L227 lo haria con el segmento alifatico de R199.

La región conectora α1-α2 esta dividida en una region no polar y otra de caracter acidico compuesta por los residuos E123, D129, D138, E140, E141. Todas las evidencias indican que esta region estaria desnaturalizada en la forma monomerica de la capside y su plegamiento probablemente requiera interacciones cuaternarias con otras subunidades y con el ARN. Algo similar ha sido observado en el TMV donde se ha encontrado que las regiones flexibles contienen gran cantidad de aminoacidos de carga negativa que se repelen entre si, evitando el ensamblaje del virus. Durante el ensamblaje y en presencia de ARN, estas regiones cambian a una conformación helicoidal que permite la estructuración del virus (Namba 2001).
DISCUSIÓN
Kendall et al. (2008) determinaron recientemente una estructura de baja resolución (22 A) del potyvirus SMV utilizando difracción de fibras, microscopia crioelectronica y de transmision. En esta reconstrucción, las subunidades de la capside se encuentran bien definidas y presentan una disposición espacial similar a la del TMV, pero con un ancho mayor y longitud menor. Las subunidades del SMV tienen 55 A de longitud y un ancho maximo de 35 A. La estructura del SMV tiene un paso de rosca de 33 A, 8,8 subunidades por vuelta y un diametro de 140 A. La densidad radial del SMV sugiere que el virion tiene un hueco central con un radio de 15 A y radio maximo de 70 A. Como puede apreciarse en la figura 5, el modelo se ajusta de manera satisfactoria a la superficie experimental y proponemos que las regiones faltantes corresponden a los extremos N y C-terminales no modelados en este trabajo. El dominio C-terminal parece ser fundamental en determinar las interacciones verticales con las helices α1 y α2. En la estructura por microscopia electronica se observa en la vista superior que cada subunidad presenta dos segmentos que forman un angulo de aproximadamente 40º. En nuestro modelo este quiebre de estructura fue obtenido de manera natural con el procedimiento de docking, lo cual sugiere que es una consecuencia del empaquetamiento de las helices del nucleo estructural.
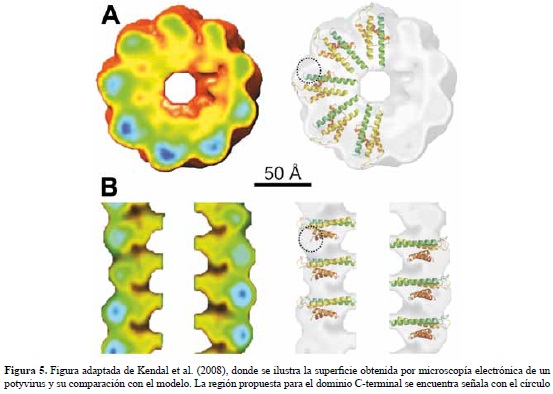
Los dos segmentos lineales estarian compuestos por α1-α2 y luego α3-α4, el dominio no modelado corresponderia a una protuberancia hacia la derecha y que proponemos que estaria formado por la union de las regiones conservadas N y C-terminales. El modelo tambien encaja de manera satisfactoria con la vista lateral de la estructura del virus. En esta imagen se observa que las subunidades de los potyvirus presentan una estructura escalonada, la parte superior corresponde a α1-α2, luego α3-α4 y por ultimo la region no modelada que determina las interacciones verticales entre subunidades. Si se asume que la region no modelada esta conformada por los residuos conservados de las regiones N y C terminales y que la forma de este dominio es aproximadamente esferico, se estima que el diametro de dicha esfera debe ser del orden de 28 A, que es un valor comparable con la regiones senaladas con circulos en la figura 5. Nemykh et al. (2008) publicaron un modelo de la estructura de la capside de PVA; sin embargo, esta estructura excede las dimensiones de potyvirus obtenidas por microscopia electronica.
En nuestra opinion, el error de dicha estructura consiste en asumir que la region C-terminal es colineal al resto de la estructura. En la propuesta aqui planteada, se sugiere que dicha region esta empaquetada con las helices α3 y α4 y tiene contactos intermoleculares con las α1 y α2 de la subunidad inmediatamente inferior.
Desafortunadamente, existen muy pocos estudios donde se evalue el efecto de mutaciones de la proteina de la capside sobre el ensamblaje de potyvirus, los cuales podrian ser de gran utilidad en el refinamiento del modelo presentado (Lin et al. 2009). Sin embargo, consideramos que la estructura presentada puede servir como una herramienta guia en el diseno de experimentos de mutagenesis dirigida que permitan la mejor comprension de los patrones de conservación de la proteina de la capside, asi como el mecanismo de ensamblaje y reconocimiento del ARN viral. Se espera que los procedimientos desarrollados en este trabajo, puedan ser empleados en el futuro proximo para modelar la estructura tridimensional de potyvirus de importancia economica en Colombia como PVY, TaLMV, PRSV, entre otros, como una estrategia para el diseno de herramientas de diagnostico (ej. diseno de anticuerpos con base en las caracteristicas estructurales de la particula viral) y de metodos de control (e. g., generación de variedades mejoradas con tolerancia/resistencia a virus) de las enfermedades asociadas en dichos virus en los agroecosistemas tropicales.
AGRADECIMIENTOS
Esta investigación se realizo gracias al apoyo económico del proyecto Colciencias: Desarrollo de metodos de detección serologica y molecular del complejo de virus asociado al mosaico del tomate de arbol en Colombia, Contrato 408-2007 y de la Dirección de Investigaciones de la Universidad Nacional de Colombia sede Medellín (DIME).
REFERENCIAS
1. Carrington JC, Freed DD, Sanders TC. 1989. Autocatalytic processing of the potyvirus helper component proteinase in Escherichia coli and in vitro. Journal of Virology 63: 4459- 4463. [ Links ]
2. Carrington JC, Dougherty WG. 1987. Small nuclear inclusion protein encoded by a plant potyvirus genome is a protease. Journal of Virology, 61: 2540-2548. [ Links ]
3. DeLano WL. 2002. The PyMOL Molecular Graphics System. DeLano Scientific Homepage. Fecha de acceso: 10 de noviembre de 2010. Disponible en: http://www.pymol.org. [ Links ]
4. Dolja VV, Haldeman R, Robertson NL, Dougherty WG, Carrington JC. 1994. Distinct functions of capsid protein in assembly and movement of Tobacco etch potyvirus in plants. EMBO Journal, 13: 1482-1491. [ Links ]
5. Doolittle RF, Garnier J, Gibrat JF, Robson B. 1996. GOR secondary structure prediction method version IV. Methods in Enzymology, 266: 540-553. [ Links ]
6. Duggal R, Hall TC. 1993. Identification of domains in Brome mosaic virus RNA-1 and coat protein necessary for specific interaction and encapsidation. Journal of Virology, 67: 6406-6412. [ Links ]
7. Ehrenfeld N, Gonzalez A, Canon P, Medina C, Perez-Acle T, Arce-Johnson P. 2008. Structure-function relationship between the tobamovirus TMV-Cg coat protein and the HR-like response. Journal of General Virology, 89: 809-817. [ Links ]
8. Hall TA. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series, 41: 95-98. [ Links ]
9. Henikoff S, Henikoff JG. 1992. Amino acid substitution matrices from protein blocks. Proceedings of the National Academy of Sciences of the United States of America, 89: 10915-10919. [ Links ]
10. Hooft RWW, Vriend G, Sander C, Abola EE. 1996. Errors in protein structures. Nature, 381: 272. [ Links ]
11. Hopp TP, Woods KR. 1981. Prediction of protein antigenic determinants from amino acid sequences. Proceedings of the National Academy of Sciences of the United States of America, 78: 3824-3828. [ Links ]
12. Kang SH, Lim WS, Hwang SH, Park JW, Choi HS, Kim KH. 2006. Importance of the C-terminal domain of Soybean mosaic virus coat protein for subunit interactions. Journal of General Virology, 87: 225-229. [ Links ]
13. Kendall A, McDonald M, Bian W, Bowles T, Baumgarten SC, Shi J, Stewart PL, Bullitt E, Gore D, Irving TC, Havens WM, Ghabrial SA, Wall JS, Stubbs G. 2008. Structure of flexible filamentous plant viruses. Journal of Virology, 82: 9546-9554. [ Links ]
14. Langenberg WG, Zhang L. 1997. Immunocytology shows the presence of Tobacco etch virus P3 protein in nuclear inclusions. Journal of Structural Biology, 118: 243-247. [ Links ]
15. Lin L, Shi Y, Luo Z, Lu Z, Zheng H, Yan F, Chen J, Chen J, Adams MJ, Wu Y. 2009. Protein-protein interactions in two potyviruses using the yeast two-hybrid system. Virus Research, 142: 36-40. [ Links ]
16. Linding R, Jensen LJ, Diella F, Bork P, Gibson TJ, Russell RB. 2003a. Protein disorder prediction: implications for structural proteomics. Structure, 11: 1453-1459. [ Links ]
17. Linding R, Russell RB, Neduva V, Gibson TJ. 2003b. GlobPlot: Exploring protein sequences for globularity and disorder. Nucleic Acids Research, 31(13): 3701-3708. [ Links ]
18. Merits A, Guo D, Saarma M. 1998. VPg, coat protein and five non-structural proteins of Potato A potyvirus bind RNA in a sequence-unspecific manner. Journal of General Virology, 79: 3123-3127. [ Links ]
19. Mink GI, Vetten HJ, Wyatt SD, Berger PH, Silbernagel MJ. 1999. Three epitopes located on the coat protein amino terminus of viruses in the Bean common mosaic potyvirus subgroup. Archives of Virology, 144: 1174-1189. [ Links ]
20. Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. 1998. Automated docking using a lamarckian genetic algorithm and empirical binding free energy function. Journal of Computational Chemistry, 19: 1639-1662. [ Links ]
21. Namba K. 2001. Roles of partly unfolded conformations in macromolecular self-assembly. Genes Cells, 6: 1-12. [ Links ]
22. Namba K, Stubbs G. 1986. Structure of Tobacco mosaic virus at 3.6 A resolution: implications for assembly. Science, 231: 1401-1406. [ Links ]
23. Nemykh MA, Efimov AV, Novikov VK, Orlov VN, Arutyunyan AM, Drachev VA, Lukashina EV, Baratova LA, Dobrov EN. 2008. One more probable structural transition in Potato virus X virions and a revised model of the virus coat protein structure. Virology, 373: 61-71. [ Links ]
24. Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. 2005. Scalable molecular dynamics with NAMD. Journal of Computational Chemistry, 26: 1781-1802. [ Links ]
25. Rajamaki M, Merits A, Rabenstein F, Andrejeva J, Paulin L, Kekarainen T, Kreuze JF, Forster RL, Valkonen JP. 1998. Biological, serological and molecular differences among isolates of a Potato A potyvirus. Phytopathology, 88: 311-321. [ Links ]
26. Riechmann JL, Lain S, Garcia JA. 1992. Highlights and prospects of potyvirus molecular biology. Journal of General Virology, 73: 1-16. [ Links ]
27. Riechmann JL, Cervera MT, Garcia JA. 1995. Processing of the Plum pox virus polyprotein at the P3-6K1 junction is not required for virus viability. Journal of General Virology, 76: 951-956. [ Links ]
28. Rost B. 2001. Review: Protein secondary structure prediction continues to rise. Journal of Structural Biology, 134: 204-218. [ Links ]
29. Rost B, Yachdav G, Liu J. 2004. The predict protein Server. Nucleic Acids Research, 32: 321-326. [ Links ]
30. Sali A, Blundell TL. 1995. Comparative protein modelling by satisfaction of spatial restraints. Journal of Molecular Biology, 234: 779-815. [ Links ]
31. Shukla DD, Ward CW. 1989. Structure of potyvirus coat proteins and its application in the taxonomy of the potyvirus group. Advances in Virus Research, 36: 273-314. [ Links ]
32. Singh RP, Valkonen JPT, Gray SM, Boonham N, Jones RAC, Kerlan C, Schubert J. 2008. Brief review: The naming of potato virus Y strains infecting potato. Archives of Virology 153: 1-13. [ Links ]
33. Urcuqui-Inchima S, Haenni AL, Bernardi F. 2001. Potyvirus proteins: a wealth of functions. Virus Research, 74 (1-2): 157-75. [ Links ]
32. Van Regenmortel MHV, Fauquet CM, Bishop DHL, Carstens EB, Estes MK, Lemon SM, Maniloff J, Mayo MA, McGeoch DJ, Pringle CR, Wickner RB. 2000. Virus Taxonomy - Seventh Report of the International Committee on Taxonomy of Viruses. Academic Press. New York. 1162 p. [ Links ]
33. Verchot J, Koonin EV, Carrington JC. 1991. The 35-kDa protein from the N-terminus of the potyviral polyprotein functions as a third virus-encoded proteinase. Virology, 185: 527-35. [ Links ]
34. Verchot J, Carrington JC. 1995. Evidence that the potyvirus P1 proteinase functions in trans as an accessory factor for genome amplification. Journal of Virology, 69: 3668-3674. [ Links ]

