










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Ciencias Químico - Farmacéuticas
Print version ISSN 0034-7418On-line version ISSN 1909-6356
Rev. colomb. cienc. quim. farm. vol.38 no.1 Bogotá Jan./June 2009
Artículo de Investigación
Aplicación de métodos modelo-dependiente y modelo-independiente en el desarrollo de una formulación de comprimidos de Captopril
Aplication of dependent and independent models for development of Captopril tablets
Gabriela Navarro1,Pablo Cabral2
1 Cátedra de Farmacotecnia, Facultad de Química, Universidad de la República, Laboratorio Farmacéutico d. n. s. ff. aa., 4876666 int. 1669, fax: 005982, Uruguay. Correo electrónico: gnavarro@fq.edu.uy
2 CIN, Facultad de Ciencias, Universidad de la República, Laboratorio Farmacéutico d. n. s. ff. aa. Uruguay. Correo electrónico: pcabral@cin.edu.uy
Recibido para evaluación: agosto 27 de 2008. Aceptado para publicación: diciembre 10 de 2008.
RESUMEN
El objetivo de este trabajo es desarrollar una formulación tecnológicamente viable de comprimidos de Captopril 25 mg cuya liberación sea comparable con la del innovador (Capoten®). Para ello se utilizaron como herramientas los perfiles de disolución in vitro. Se diseñaron dos formulaciones (F1 y F2) que contenían dicho ingrediente activo para ser procesadas por el método de compresión directa. En cada caso se evaluaron las características de las mezclas obtenidas como modo de determinar su viabilidad tecnológica, resultando F1 y F2 aptas para la compresión. Para ambas formulaciones, los parámetros fisicoquímicos de los comprimidos se encontraron dentro de especificaciones. Se compararon los perfiles de disolución de F1 y F2 con los del innovador en tres medios de diferente pH, aplicando métodos modelo- dependientes e independientes. Los perfiles de disolución in vitro de F2 fueron similares a los del innovador en los medios estudiados; por tanto, F2 presentó una liberación in vitro comparable con Capoten®
Palabras clave: Captopril, tecnología, perfil de disolución, modelos.
ABSTRACT
The aim of this work is the development of a technologically viable tablet formulation of Captopril 25 mg tablets with a comparable release with the original (Capoten®). The used tools were in vitro dissolution profiles. Two formulations for direct compression (F1 and F2) containing the active ingredient were designed. The characteristics for both mixtures were studied and technological viability determinated, resulting F1 and F2 suitable for compression. Both tablets formulations presented physicochemical parameters within specifications. Dissolution profiles of F1 and F2 were compared with the original's using three different pH, model-dependent and model-independent methods were applied for that. The in vitro dissolution profiles of F2 were similar to those of the innovator in the studied media, therefore F2 showed a comparable in vitro release with Capoten®.
Key words: Captopril, technology, dissolution profile, models.
INTRODUCCION
Desarrollar nuevas formulaciones involucra aplicar principios biofarmacéuticos y tecnológicos con el objetivo de diseñar un sistema óptimo, seguro y eficaz de liberación de un ingrediente activo desde una forma farmacéutica.
Establecida la dosis del activo en función de su comportamiento farmacocinético y de su objetivo terapéutico, se deben prever los siguientes aspectos:
1. Caracterización del ingrediente activo: desde los puntos de vista organoléptico (sabor, color, aroma), físico (aspecto, estructura cristalina o amorfa, granulometría, densidad aparente, solubilidad, higroscopicidad, punto de fusión), farmacotécnico (reología, plasticidad o elasticidad, compresibilidad), químico (estabilidad a la luz, aire, calor, humedad), de incompatibilidades, farmacocinético (biodisponibilidad, vida media de eliminación, metabolitos activos) y microbiológico (grado y tipo de contaminación microbiana). La elección de los excipientes que se han de utilizar está dada por su compatibilidad con el ingrediente activo o entre los diferentes componentes entre sí. El estudio de posibles interacciones se lleva a cabo con mezclas binarias del activo y cada uno de los componentes en condiciones forzadas de luz, temperatura, humedad, medio ácido, alcalino u oxidante.
2. Tecnología de fabricación y elección del método de compresión. La absorción del fármaco liberado desde un comprimido con posterioridad a su administración oral depende de su liberación desde la forma farmacéutica, de su velocidad de disolución en las condiciones fisiológicas y de su permeabilidad en el tracto gastrointestinal. A causa de la naturaleza crítica del proceso de disolución, evaluar la disolución in vitro del activo puede ser relevante para predecir el desempeño del fármaco in vivo.
Ahora bien, los estudios de disolución in vitro para comprimidos de liberación convencional se utilizan como guía para desarrollar nuevas formulaciones e instrumentos de aseguramiento de la calidad para medir la uniformidad lote a lote (2).
Para desarrollar nuevas formulaciones de genéricos, la fda propone para la gran mayoría de los fármacos llevar a cabo estudios de bioequivalencia in vivo entre la formulación en estudio y el innovador que se toma como referencia (3).
El objetivo de este trabajo es diseñar una formulación tecnológicamente viable de Captopril 25 mg, cuya liberación in vitro sea comparable con la de la referencia (Capoten ®) como modo de aproximación a la bioequivalencia. La herramienta utilizada para ello es la comparación de los perfiles de disolución in vitro aplicando métodos modelodependientes e independientes.
MATERIALES METODOS
Materiales y equipos
Máquina de comprimir rotativa (Manesty D3B), con punzones de 6 mm planos.
Mezcladora de cintas (Werner & Pfleiderer).
Durómetro (Erwecka TBH 20).
Equipo de disolución PROLABO Dissolutest 07 170.402.
Cromatógrafo de gran desempeño, bomba (Waters 600E), detector de arreglo de diodos (Waters 991).
Espectrofotómetro uv-Visible (Spectronic 1201).
Reactivos calidad analítica.
Las materias primas cumplen con la calidad requerida para su empleo según USP 31 (4).
Métodos
Formulación y estudios de compatibilidad
Dadas las características físicas del principio activo, la dosis requerida y la disponibilidad de excipientes con buenas características de flujo y compresibilidad (4, 5, 6), se elige la compresión directa como método de obtención de los comprimidos y se diseñan dos formulaciones cuyas composiciones difieren en la relación lactosa/celulosa microcristalina.
Las formulaciones propuestas se muestran en la Tabla 1.

Los estudios de compatibilidad se llevaron a cabo realizando mezclas binarias de Captopril y cada uno de los excipientes seleccionados, sometiéndolas a 40°C y 70% de humedad durante 30 días, para determinar luego la presencia de productos de degradación mediante técnicas de cromatografía de excelente desempeño (HPLC, por sus siglas en inglés).
Obtención de los comprimidos y estudios realizados
En cada caso las mezclas obtenidas se caracterizaron desde el punto de vista tecnológico y fisicoquímico y se determinaron los siguientes parámetros: ángulo de reposo, densidad aparente, densidad compactada, índice de compresibilidad, porosidad, contenido de humedad, índice de dureza-friabilidad y uniformidad de dosis (3, 5, 8). A los comprimidos obtenidos en cada proceso se les determinaron las propiedades físico- mecánicas y tecnológicas: dimensiones, dureza y friabilidad (3, 6). Así mismo, se les realizaron los correspondientes análisis fisicoquímicos: dosificación, uniformidad de contenido y porcentaje de disolución (4).
Comparación de los perfiles de disolución
El ensayo de disolución se llevó a cabo sobre seis comprimidos, para cada formulación y referencia, utilizando aparato 1 (4) (50 rpm), temperatura 37 ± 1 °C.
La cuantificación del fármaco liberado en cada medio a 1; 2,5; 4; 5; 10 y 15 minutos se realizó por espectrofotometría UV a 205 nm, cuya técnica fue previamente validada.
Los perfiles de disolución de los comprimidos de cada formulación se compararon con el de la referencia en tres medios a pH 1,0 (ácido clorhídrico 0,1 N), pH 5,5 (agua destilada) y pH 7,4 (buffer fosfato) y se promediaron los porcentajes de disolución obtenidos para cada tiempo.
La comparación de los perfiles de disolución se realizó utilizando modelos independientes y dependientes, de modo de poder evaluar la similitud in vitro de dichos perfiles entre la referencia y las fórmulas propuestas.
Aplicación del método modelo-independiente
En este caso se utilizaron las aproximaciones de los factores de diferencia f1 y similitud f2 (9).
El factor de diferencia f1 calcula el porcentaje diferencial entre las dos curvas a cada tiempo, y es una medida relativa del error entre dos curvas. Se calcula según la fórmula: ecu1

El factor de f2 , similitud, es una transformación logarítmica del recíproco de la raíz cuadrada de la suma de los cuadrados de los errores, y es una medida de la semejanza en el porcentaje de disolución entre dos curvas. ecu2

Para asegurar similitud o equivalencia entre dos curvas, los valores de f1 deben ser menores a 15 (0 a 15), y los de f2 , mayores a 50 (50 a 100) (9).
Aplicación del método modelo-dependiente
Dentro de los distintos modelos matemáticos descritos para ajustar los perfiles de disolución, se aplicaron los siguientes: primer orden, Higuchi, raíz cúbica, Weibull y orden cero a los perfiles de las dos fórmulas propuestas y la referencia en los tres medios estudiados (10).
Una vez determinado el modelo de mejor ajuste, se calcularon los parámetros constante de disolución y velocidad máxima de disolución en todos los casos y se los comparó estadísticamente aplicando un nivel de significación α = 0,05.
RESULTADOS Y DISCUSIONFormulación y estudios de compatibilidad
Los análisis de las mezclas binarias del activo y excipientes no demostraron presencia de productos de degradación que denotaran incompatibilidades entre sí aplicando técnicas de HPLC. Es decir, no presentaban picos secundarios al principal, y la dosificación del activo no mostró cambios significativos durante el estudio.
Caracterización tecnológica y fisicoquímica de las mezclas
En la Tabla 2 se muestran los parámetros tecnológicos y fisicoquímicos determinados de las mezclas correspondientes a las formulaciones F1 y F2.

Los parámetros tecnológicos en las dos mezclas evaluaron sus características de flujo y compresibilidad con objeto de poder predecir si eran adecuadas para obtener comprimidos en el equipo de compresión. Se debe tener en cuenta que dicha obtención se realiza mediante el llenado de matrices por volumen y a gran velocidad, de modo que para asegurar la uniformidad de llenado de dichas matrices, que asegure la uniformidad de peso y con ello la uniformidad de dosis (con la previa determinación de uniformidad de mezclado), es crítico el hecho de que la mezcla tenga un flujo constante y uniforme. Así mismo debe tener buenas características de compresibilidad que permitan formar el comprimido.
En ambos casos los valores obtenidos indicaron buen flujo y compresibilidad de los lechos de polvo.
La uniformidad de dosis en la mezcla permitió corroborar el tiempo óptimo de mezclado y asegurar una distribución homogénea del activo en el lecho, con lo que se logra uniformidad de dosis en los comprimidos que constituyen la unidad posológica. Dicho parámetro se encontró dentro de especificaciones para F1 y F2.
Caracterización tecnológica y fisicoquímica de los comprimidos
Las variables tecnológicas y fisicoquímicas determinadas para los comprimidos correspondientes a las dos formulaciones propuestas se describen en Tabla 3.

La determinación de dimensiones, dureza y friabilidad evalúan las características físicas de lo comprimidos, la resistencia a la rotura y abrasión, las cuales deben ser adecuadas para permitir su posterior manipulación durante el fraccionamiento industrial y utilización de parte del paciente.
Las propiedades fisicomecánicas determinadas para los comprimidos obtenidos de las fórmulas F1 y F2 indicaron que en los dos casos se obtuvieron comprimidos de dimensiones similares, con buena resistencia mecánica tanto a la abrasión como a la rotura.
La dosificación, determinación de uniformidad de contenido y porcentaje de disolución aseguran la presencia del fármaco en los valores declarados, su adecuada distribución en todo el lote y su porcentaje de disolución in vitro en el tiempo especificado (4).
Dichos parámetros, además de la friabilidad, se hallaron dentro de especificaciones de la UPS 31, tanto para F1 como para F2.
Comparación de perfiles de disolución
Las Figuras 1, 2 y 3 muestran los perfiles de disolución de F1 y F2 y la referencia en los tres medios estudiados en ellos; se verificó, para los tres medios, una rápida disolución, tanto de los comprimidos de referencia como de los correspondientes a las fórmulas propuestas.
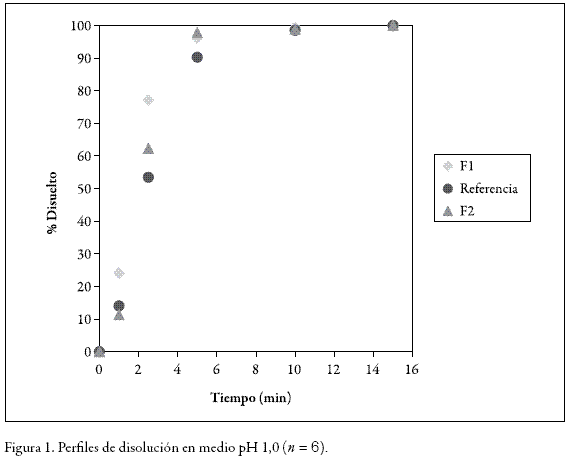


En los tres medios, durante los primeros minutos, los valores de los comprimidos de F2 estuvieron siempre más próximos a los de la referencia que los de F1.
Aplicación del método modelo-independiente
En la Tabla 4 se muestra la comparación de los perfiles de disolución en los distintos medios de los comprimidos de cada fórmula con la referencia, al aplicar los factores F1 y F2.

La comparación de los perfiles de disolución promedio de las formulaciones F1 y F2 respecto a la referencia, aplicando el método modelo-independiente (Tabla 3), indicó que para F1 se detectan diferencias significativas en dichos perfiles, respecto al innovador en los tres medios estudiados en virtud de que los valores del factor f1 son mayores a 15. Al evaluar el factor f2 se confirmó la falta de similitud entre las curvas, ya que los valores obtenidos son menores a 50 en todos los medios.
En el caso de los perfiles de los comprimidos F2, en los que la relación lactosa/celulosa microcristalina fue menor a 1, se puede afirmar que existe similitud con la referencia aplicando el mismo modelo, ya que los valores de f1 fueron menores a 15, y los de f2, mayores a 50 en los tres medios.
Aplicación del método modelo-dependiente
El modelo matemático que mejor se ajustó a los perfiles de disolución de los comprimidos fue el de orden 1 para la referencia F1 y F2 (9).
Aplicando dicho modelo, se determinaron las constantes de disolución y la velocidad máxima de disolución para los comprimidos de las dos formulaciones propuestas en cada medio; dichos resultados se muestran en las Tablas 5, 6 y 7.



En estas tablas se observan los valores obtenidos de k de disolución y velocidad máxima de disolución para la referencia, F1 y F2 a pH 1,0, 5,5 y 7,4, respectivamente, aplicando el modelo de primer orden.
La comparación de las constantes de disolución y velocidad máxima de disolución para F2 y la referencia se realizó estadísticamente aplicando un nivel de significancia, α =0,05, las cuales no mostraron diferencias significativas en ninguno de los tres medios.
En el caso de los comprimidos de fórmula F1, dichos parámetros no fueron estadísticamente comparables con la referencia, lo cual concuerda con los resultados obtenidos aplicando un modelo independiente.
Se observó que el aumento de la proporción de celulosa microcristalina causó disminución en la velocidad de disolución del fármaco, lo que se reflejó en los valores de las k de disolución de cada formulación.
CONCLUSIONES
Las formulaciones F1 y F2 son tecnológicamente viables para obtener comprimidos de Captopril 25 mg mediante el proceso de compresión directa, que cumple con la farmacopea americana, por lo que con ambas formulaciones se obtienen comprimidos aptos para su aplicación terapéutica. Los comprimidos obtenidos a partir de la formulación F2 presentan parámetros de liberación in vitro comparables a los de la referencia en todos los medios estudiados. Se demostró que la relación lactosa/celulosa microcristalina influye en la cinética de disolución in vitro del Captopril.
REFERENCIAS
1. J. Flórez, "Farmacología humana", Masson Multimedia Eds., pp. 344 a 353. [ Links ]
2. G. Banker y C. Rhodes. C "Drugs and the Pharmaceutical Sciences". En: "Modern pharmaceutics", Marcel Dekker inc., New York. [ Links ]
3. FDA, Center for Drug Evaluation and Research (CDER), Guidance for Industry: "Waiver of in vivo bioavailability an bioequivalence studies for immediate- release oral dosage forms based on a biopharmaceutics classification system", August 2000. [ Links ]
4. United States Pharmacopeia, USP 31, Inc. Rockville. [ Links ]
5. H. Lieberman y L. Lachman. En: "Pharmaceutical Dosage Forms"; Ed. Marcel Dekker, inc., New York, 1981, Vol. 2, pp. 185-262. [ Links ]
6. Remington, "The science and practice of pharmacy", Mack Printing Company, Pennsylvania, 1995, Vols. 1 y 2. [ Links ]
7. A. Kibbe. En: "Handbook of pharmaceutical excipients", American Pharmaceutical Association, Washington, D. C., Pharmaceutical Press, London, 2000. [ Links ]
8. M. Campo, I. Perdomo, A. Iraizoz y M. Rodríguez, Rev. Cubana Farm., 35, 165 (2001). [ Links ]
9. FDA, Center for Drug Evaluation and Research (CDER), "Guidance for Industry: Dissolution Testing of Immediate Release Solid Oral Dosage Forms", 1997. [ Links ]
10. A. Gordon y B. Marival, software "Biofarmacia moderna", TSRL Inc. Eds., Michigan, 2003. [ Links ]

