











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkLa hepatitis B constituye una de las hepatitis virales más importantes en salud pública por estar relacionada en su fase crónica con el desarrollo de cirrosis y carcinoma hepatocelular 1. La proporción de individuos adultos cuya infección aguda por HBV progresa a la fase crónica es de 5 a 10 %, en tanto que en niños menores de cinco años la proporción es de 25 a 30% y, en recién nacidos, hasta de 90 % 2.
A nivel mundial, se estima que, aproximadamente, 240 millones de personas presentan infección crónica y más de 686.000 mueren cada año como consecuencia de la enfermedad y sus complicaciones 3. La seroprevalencia de hepatitis B en Latinoamérica es de 0,48 %, aproximadamente, y en Colombia se estima en 2,29 % 4. En el 2016, se notificaron 1.967 casos de hepatitis B al sistema nacional de vigilancia epidemiológica (Sivigila), 42 % de los cuales provenía de Antioquia, Bogotá y Norte de Santander; la incidencia nacional de hepatitis B fue de cuatro casos por cada 100.000 habitantes 5. Con un patrón epidemiológico heterogéneo debido a las diferencias geográficas, étnicas, culturales y socioeconómicas, se considera que el país tiene una endemia de baja a intermedia 6, aunque en algunos departamentos, entre ellos Amazonas, Chocó, Guaviare, Magdalena y Norte de Santander, es alta 7-9.
El virus de la hepatitis B (HBV) pertenece a la familia Hepadnaviridae, género Orthohepadnavirus, su genoma es una molécula de ADN circular, parcialmente de doble hebra y de 3,2 kb, aproximadamente, constituido por marcos de lectura parcialmente superpuestos que codifican para las proteínas preCore/Core, preS1/preS2/S, polimerasa y X 10. El análisis filogenético de las cepas del HBV circulantes a nivel mundial ha permitido la identificación de 10 genotipos (A-J) con una divergencia en la secuencia genómica mayor de 8 %, y la asignación de más de 30 subgenotipos definidos como aquellos linajes conformados por cepas con una divergencia de nucleótidos de 4 a 8 % 11,12.
En Colombia, se ha reportado la circulación de los genotipos y subgenotipos A1, A2, C, D3, E, F1a, F1b, F3 y G 6,8,13-19, siendo el genotipo F el de mayor prevalencia en Suramérica 12.
Aunque la progresión y la gravedad de la hepatitis B responden a múltiples factores, los estudios epidemiológicos han permitido establecer asociaciones entre los diferentes genotipos y variables tales como las vías de transmisión, la progresión a la fase crónica de la enfermedad, el tiempo de progresión y la gravedad de la enfermedad 20.
Por otro lado, diversos estudios han permitido la identificación de mutaciones puntuales en el dominio de la transcriptasa inversa (reverse transcriptase, RT) del HBV que confieren resistencia a los principales medicamentos antivirales disponibles: lamivudina (3TC), entecavir (ETV), telbivudina (LdT), adefovir (ADV) y tenofovir (TDF) 21. Asimismo, se han identificado mutaciones puntuales y deleciones en las regiones S, preS1 y preS2 del gen de la envoltura del HBV, las cuales se han asociado con la presentación clínica de la enfermedad 22, con la neutralización a cargo de anticuerpos generados tras la vacunación o como resultado de una infección natural 23, o de casos de infección oculta por HBV 24, así como con la sensibilidad de los inmunoensayos utilizados para el diagnóstico serológico 25.
En el presente estudio, se llevó a cabo la detección molecular, la genotipificación y la identificación de mutaciones en las regiones codificantes del antígeno S y el dominio RT de la polimerasa del HBV a partir de 495 muestras de suero con resultado positivo para HBsAg provenientes de los 32 departamentos de Colombia, las cuales se recibieron para diagnostico de hepatitis B en el Laboratorio de Virología del Instituto Nacional de Salud, durante el periodo 2002-2014.
Materiales y métodos
Muestras clínicas
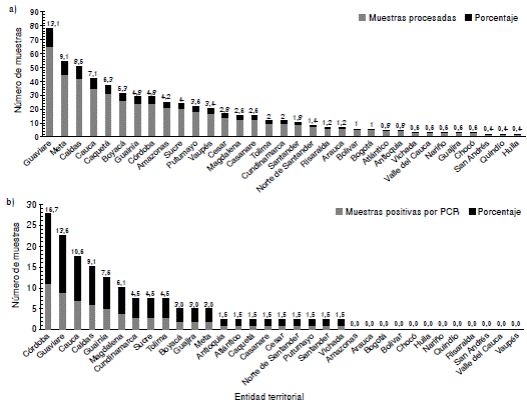
Se hizo un estudio descriptivo, longitudinal y retrospectivo, de 495 muestras de sueros provenientes de 32 departamentos del país y de Bogotá, conservadas en congelación entre los -20 °C y los -30 °C en la seroteca del Grupo de Virología del Instituto Nacional de Salud para el diagnóstico de hepatitis B, y recolectadas durante el periodo 2002-2014 (figura 1a). Se seleccionaron muestras con resultado positivo en la prueba ELISA para el antígeno de superficie de HBV (HBsAg), no necesariamente confirmado mediante prueba de neutralización.
Extracción del ADN viral y amplificación de las regiones codificantes del antígeno S y del dominio RT de la polimerasa del HBV mediante PCR
Para la extracción del ADN viral, y ante la eventual necesidad de detectar virus con genoma de ARN (virus de hepatitis A, C, delta y E), se utilizó el estuche PureLink Viral RNA/DNA Mini Kit™ (Thermo Fisher Scientific Inc., Waltham, MA, USA), siguiendo las instrucciones del fabricante.
Para la amplificación y secuenciación de las regiones codificantes del antígeno S y del dominio RT de la polimerasa del HBV, se utilizaron los oligonucleótidos externos HBPr1_Sense 5’-gggt caccatattcttggg-3’ y HBPr374_Antisense 5’-gttcc gcagtatggatcggcagagg-3’, previamente descritos por Stuyver, et al.26, los cuales se extienden desde la posición 2816-3182...1-1281 del genoma de referencia, número de acceso en GenBank:NC_003977.2.
La mezcla de reacción para la PCR incluyó 10 pmoles de cada oligonucleótido, 1,35 U de Platinum Taq DNA Polymerase High Fidelity™ (Thermo Fisher Scientific Inc., Waltham, MA, USA), 60 mM de Tris-SO4 (pH 8,9), 18 mM de (NH4)2SO4, 2,4 mM de MgSO4, 480 nM de cada dNTP, 5 μl del extracto de ADN viral y agua libre de nucleasas, para un volumen final de 25 μl.
El perfil térmico consistió en una desnaturalización inicial a 94 °C durante dos minutos, seguida de 35 ciclos de amplificación (a 94 °C durante 30 segundos, a 50 °C durante 30 segundos, a 72 °C durante dos minutos) y una extensión final a 72 °C durante cinco minutos.
Los amplicones esperados, de 1.648 pb, aproximadamente, se separaron mediante electroforesis en gel de agarosa al 1 % en TAE 1X, se tiñeron con SYBR Safe DNA Gel Stain™ (Thermo Fisher Scientific Inc., Waltham, MA, USA) y se visualizaron mediante el fotodocumentador GelDoc XR™ (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Secuenciación, edición de secuencias y análisis filogenético
Los productos de PCR positivos para ADN del HBV se purificaron utilizando el estuche QIAquick PCR Purification Kit™ (QIAGEN Inc, Valencia, CA, USA) y se secuenciaron usando el estuche BigDye Terminator, versión 3.1, Cycle Sequencing Kit™ (Thermo Fisher Scientific Inc., Waltham, MA, USA), según las instrucciones del fabricante.
La secuenciación se llevó a cabo en un Genetic Analyzer ABI 3130™ (Applied Biosystems, Foster City, California, USA). Las secuencias obtenidas se visualizaron, editaron y ensamblaron mediante el módulo SeqMan de la suite Lasergene, versión 7.2.1™ (DNASTAR Inc., Madison, WI, USA) y, posteriormente, se alinearon empleando el algoritmo ClustalW con el programa MEGA 6.0 27. Se incluyeron secuencias disponibles en el GenBank (http://www.ncbi.nlm.nih.gov/genbank) pertenecientes a los diferentes genotipos y subtipos del HBV humano, así como el máximo número de secuencias colombianas previamente reportadas y con la longitud genómica requerida para estos análisis y, además, una secuencia de HBV derivada del mono lanudo como grupo externo (número de acceso en GenBank: AY226578).
Para la reconstrucción del árbol filogenético, se generaron dos matrices: la primera, para analizar la longitud máxima del genoma, se extendía entre las posiciones 2858-3182...1-1266 (1.591 pb) y la segunda, para incluir el máximo número de secuencias colombianas previamente reportadas, se extendía entre las posiciones 281 y 657 (377 pb) del genoma circular de referencia NC_003977.2. Para cada matriz se seleccionó el modelo evolutivo que mejor se ajustara a los datos, utilizando el programa jModelTest 28.
El análisis filogenético se hizo con el programa MEGA 6.0 y el método de neighbor-joining. El soporte de la topología del árbol se obtuvo mediante un proceso de bootstrap de 1.000 réplicas.
Análisis de mutaciones de resistencia a medicamentos antivirales y de escape
Para la determinación del impacto de las diferentes mutaciones presentes en las secuencias del HBV circulantes en el país, se emplearon dos herramientas bioinformáticas disponibles en línea: la HIV-grade HBV drug resistance interpretation (DRI) 29 y la Geno2pheno [HBV] (G2P) 30, las cuales permiten determinar el genotipo viral, identificar las mutaciones en la región codificante del dominio RT de la polimerasa viral asociadas con resistencia a fármacos e identificar mutaciones en el marco de lectura abierto (Open Reading Frame, ORF) del gen que codifica para el antígeno S pequeño (S-HBsAg), previamente asociadas con escape por disminución de la unión al anticuerpo.
La DRI y la G2P se encuentran disponibles en los enlaces: http://www.hiv-grade.de/hbv_grade/deployed/grade.pl?program=hbvalg y http://hbv.geno2pheno.org/, respectivamente.
Resultados
Distribución de las muestras por departamento
En el estudio se incluyeron 495 muestras de sueros con resultado positivo para HBsAg, lo cual permitió tener una representación de las diferentes regiones de Colombia (figura 1a), con un mínimo de dos y un máximo de 65 sueros provenientes de cada una de las 33 entidades territoriales, incluida Bogotá. Sin embargo, la incidencia del HBV reportada por año epidemiológico por las distintas entidades territoriales, es diferente al número de muestras recibidas en el Instituto Nacional de Salud, ya que la remisión de muestras depende de la capacidad de diagnóstico de cada departamento y del nivel de referencia que registran frente al Instituto.
Caracterización molecular de genotipos y subgenotipos circulantes del HBV en Colombia
Mediante amplificación por PCR convencional de 66 muestras (13,3 % del total de las muestras procesadas) provenientes de 21 unidades territoriales del país, se obtuvo un fragmento de banda única y del tamaño esperado correspondiente al ORF S y, región codificante del dominio RT de la polimerasa del HBV. Los departamentos de Córdoba, Guaviare, Cauca, Caldas, Guainía y Magdalena, presentaron la mayor positividad, contribuyendo en conjunto con el 63,6 % de las muestras positivas mediante detección molecular (figura 1b).
Del total de muestras amplificadas por PCR, 28 (42,4 %) se secuenciaron y, mediante el análisis filogenético, se identificaron los genotipos A y F, cuyos subgenotipos fueron el A2 y el F3, respectivamente (figura 2). De las 28 secuencias, 24 (85,7 %) correspondieron al genotipo F y 4 (14,3 %) al genotipo A, similar a lo reportado para el país en estudios previos 7,13,19. El análisis filogenético mediante la matriz 1 con secuencias de longitud de 1.591 pb, permitió la identificación confiable de los subgenotipos.

Figura 2 Inferencia filogenética por neighbor-joining (NJ) del HBV, basada en el gen de la polimerasa viral (parcial de 1.591 pb). Se utilizó el método de NJ con 1.000 réplicas en el proceso de bootstrap y el modelo de sustitución de nucleótidos K2P+G (Shape: 0,77). La secuencia del virus de la hepatitis B del mono lanudo (número de acceso en GenBank: AY226578) se utilizó como grupo externo solo con fines de visualización. Los rombos rojos representan las cepas colombianas secuenciadas en el presente estudio y, los negros, las cepas colombianas previamente depositadas en GenBank.
Todas las secuencias de HBV de genotipo F identificadas en el presente estudio, pertenecían al subgenotipo F3 con una gran confiabilidad (soporte en el proceso de bootstrap de 92 %). De igual forma, todas las secuencias del HBV, genotipo A, identificadas pertenecían al subgenotipo A2, con una gran confiabilidad (soporte en el proceso de bootstrap de 99 %), similar a lo previamente reportado 16.
Con base en la región genómica representada por la matriz 1, cuyos sitios variables se muestran en la figura 3, se estimó la divergencia evolutiva promedio del HBV entre cada par de cepas colombianas de los subgenotipos A2 (0,003 sustituciones por sitio) y F3 (0,007 sustituciones por sitio) y entre dichos subgenotipos (0,162 sustituciones por sitio entre cada par de cepas colombianas). No se identificaron secuencias pertenecientes a los genotipos C, D, E y G, que previamente habían sido reportados en el país 16.

Figura 3 Variabilidad genética en la región secuenciada correspondiente al gen de la polimerasa (parcial) del HBV. Se alinearon las secuencias de 1.591 pb del HBV en muestras del periodo 2002-2014 obtenidas en el presente estudio, frente a la secuencia de referencia NC_003977. La región secuenciada se extiende entre los nucleótidos 2858-3182 y 1-1266 del genoma circular de referencia. Los sitios resaltados en negro corresponden a posiciones con variabilidad genética en el individuo. M: C/A; R: A/G; Y: C/T; W: A/T
Teniendo en cuenta que las secuencias disponibles pertenecientes a dichos genotipos no fueron lo suficientemente extensas o no correspondían completamente a la región secuenciada en el presente estudio (matriz 1), se generó una segunda matriz (matriz 2), con el fin de establecer las relaciones filogenéticas y obtener un panorama completo de los genotipos que han circulado en el país (figura 4). En este árbol filogenético se pueden observar los diferentes genotipos representados; sin embargo, en general, no fue posible establecer subgenotipos debido a los bajos soportes en el bootstrap, resultantes del análisis de un fragmento corto con una menor cantidad de sitios informativos a nivel filogenético.

Figura 4 Genotipos del HBV presentes en Colombia. Inferencia filogenética por neighbor-joining basada en un fragmento de 377 pb (posiciones 1.155-1.531 del genoma de referencia NC_003977). Se utilizaron 1.000 réplicas de bootstrap y el modelo de sustitución de nucleótidos K2P+G (Shape: 0,23). La secuencia del HBV del mono lanudo (número de acceso en GenBank: AY226578) se utilizó como grupo externo con fines de visualización. Los rombos rojos representan las cepas colombianas secuenciadas en el presente estudio y, los negros, las cepas colombianas previamente depositadas en GenBank.
La secuencia de la muestra identificada con el código 49791_Cauca_CO_2003, presentó un gran número de sitios variables, los cuales se designaron según el código internacional de ambigüedad de nucleótidos (figura 3). La gran frecuencia de sustituciones no sinónimas en dicha secuencia (figura 5), sugiere la presencia de una infección simultánea con dos diferentes cepas del HBV; sin embargo, sería necesaria la clonación y secuenciación de los clones del producto de la PCR para confirmar o refutar dicha hipótesis.

Figura 5 Variación de nucleótidos y presencia de mutaciones de resistencia en el dominio RT del gen que codifica para la transcriptasa inversa del HBV. Alineamiento de secuencias del HBV presente en Colombia en el periodo 2002-2014 con respecto a la secuencia de referencia NC_003977. Los sitios resaltados en negro corresponden a posiciones con variabilidad en el individuo. Los sitios resaltados en amarillo corresponden a mutaciones de resistencia a medicamentos antivirales. En el alineamiento solo se presentan los sitios variables.
Mutaciones en el ORF S y la región codificante del dominio RT de la polimerasa del HBV circulante en Colombia y su asociación con resistencia a medicamentos antivirales y con el escape
Mediante el uso de herramientas bioinformáticas, se identificaron mutaciones asociadas con resistencia a medicamentos antivirales en dos de las muestras secuenciadas. La secuencia de la cepa 49884_Cundinamarca_CO_2003, perteneciente al genotipo A, presentó simultáneamente dos sustituciones (L180M y M204V) previamente asociadas con resistencia a LdT, 3TC y ETV 31,32, e identificadas recientemente en un paciente colombiano con hepatopatía en estado terminal 33 (figura 5). La secuencia de la cepa 60265_Cesar_CO_2004, perteneciente al genotipo F, presentó la sustitución I169L asociada con resistencia a ETV. De todas maneras, en los artículos pertinentes se señala la necesidad de acopiar mayor evidencia clínica sobre dicha asociación 34.
En cuanto a la identificación de mutaciones en el S-HBsAg, únicamente la secuencia de la cepa 48877_Guaviare_CO_2002, perteneciente al genotipo F, presentó la mutación de escape P120Q (figura 6). Las tres mutaciones asociadas con la resistencia a los medicamentos antivirales, se identificaron en muestras provenientes de hombres con edades entre los 17 (dos mutaciones) y los 51 años, en tanto que la mutación de escape se identificó en una mujer de 22 años, cuyo antecedente de vacunación no se pudo determinar (cuadro 1).

Figura 6 Variación de aminoácidos y presencia de mutaciones de escape en el antígeno S pequeño del HBV (S-HBsAg). Alineamiento de las secuencias del HBV presente en Colombia en el periodo 2002-2014 con respecto a la secuencia de referencia traducida NC_003977. Los sitios resaltados en negro corresponden a posiciones con variabilidad en el individuo. El sitio resaltado en amarillo corresponde a una mutación de escape. En el alineamiento solo se presentan los sitios variables. *: codón de parada
Cuadro 1 Mutaciones de resistencia y escape en los genes P y S del HBV, Colombia, 2002-2014

* Código interno del Laboratorio de Virología del Instituto Nacional de Salud HBV-DRI: HIV-grade HBV drug resistance interpretation M: masculino; F: femenino; LdT: telbivudina; 3TC: lamivudina; ETV: entecavir; FTC: emtricitabine
Dos de las secuencias del HBV obtenidas en el presente estudio, presentaron una deleción en la región preS1-preS2 (figura 7) y, aunque dicha deleción no involucró el antígeno S pequeño (S-HBsAg), sí comprometía 15 codones de la región codificante del antígeno S mediano (M-HBsAg), incluido el codón de inicio, así como 35 codones codificantes del antígeno S grande (L-HBsAg).

Figura 7 Región codificante de los antígenos de superficie del HBV. a) Representación gráfica de los antígenos de superficie grande (L-HBsAg), mediano (M-HBsAg) y pequeño (S-HBsAg) del HBV. b) Delimitación de la región correspondiente a la deleción presente en dos cepas del HBV colombianas. Las posiciones genómicas se calcularon con base en el genoma de referencia del HBV, número de acceso en GenBank NC_003977.2. La región delimitada con signo de interrogación no se pudo resolver en los electroferogramas obtenidos.
Discusión
El país presenta un patrón heterogéneo en la incidencia y prevalencia de hepatitis B. En el 2016 los departamentos de Guaviare, Amazonas y Vaupés registraron la mayor incidencia de casos (12-20/100.000 habitantes) en tanto que Boyacá, Cauca, San Andrés y Buenaventura notificaron la incidencia mas baja (1-3/100.000 habitantes) 5. Históricamente, también se ha estimado un elevado subregistro, cercano al 80 %, debido al bajo porcentaje de notificación en ciertas unidades territoriales 35,36, lo cual permite inferir cifras mayores de las reportadas oficialmente.
Por tratarse de una enfermedad que en una elevada proporción de los individuos infectados se desarrolla de forma crónica y silenciosa, y sin una fase aguda de mayor consideración, la carga real de la enfermedad y la magnitud de este problema de salud pública solo llegan a ser evidentes a medida que progresa el daño hepático y se manifiesta como cirrosis o carcinoma hepatocelular, condiciones potencialmente mortales que requieren trasplante hepático y dan una reducida esperanza de vida 37,38. En el 2016, se reportaron al Sivigila 38 muertes probables por hepatitis B 5. A pesar de la lucha para controlar la transmisión mediante la aplicación de una vacuna efectiva en la población expuesta y de la promoción de conocimientos, actitudes y prácticas que minimicen los factores de riesgo, esta enfermedad continúa siendo un problema para los nacidos antes de 1992 y para quienes no han sido vacunados, que, además, se convierten en una importante fuente de infección y transmisión.
El principal objetivo del presente estudio fue la caracterización molecular del HBV a partir de muestras de pacientes remitidas para diagnóstico y captadas mediante el sistema de vigilancia nacional por laboratorio de las hepatitis virales. A pesar de que la PCR fue optimizada y la utilización de controles positivos permitió demostrar su correcto funcionamiento en todos los ensayos, el porcentaje de detección observado fue relativamente bajo (13,3 %) debido a la inclusión de muestras sin resultado confirmatorio del HBsAg mediante ensayo de neutralización; está documentado que existe un porcentaje de falsos positivos en los ensayos mediante la técnica ELISA debido a la unión inespecífica de antígenos. En 28 (42,4%) de las 66 muestras amplificadas, se lograron la secuenciación y el ensamblaje de la región genómica esperada. A pesar de que algunas muestras lograron amplificarse y secuenciarse parcialmente, se excluyeron por carecer de la región completa objeto del análisis, o por arrojar electroferogramas de baja resolución que impedían su correcto ensamblaje.
Los análisis filogenéticos del HBV han sido relevantes desde el punto de vista epidemiológico y clínico, pues han permitido rastrear el origen ancestral y reciente de los virus, estimar el tiempo de aparición de los diferentes genotipos y subgenotipos, establecer asociaciones entre los genotipos, las diferentes vías de transmisión y las manifestaciones clínicas de la enfermedad, y predecir la respuesta al tratamiento antiviral 21,39. Además, se han convertido en un complemento importante en el estudio de brotes para establecer la cadena de transmisión 40.
En el continente americano circula predominantemente el genotipo F; la información filogenética sugiere que este genotipo se originó en poblaciones amerindias de Norteamérica, Centroamérica y Suramérica hace 8.900 años, aproximadamente 42,43. El genotipo F se ha relacionado con una mayor frecuencia de carcinoma hepatocelular y muerte 44,45. Este genotipo se ha venido detectando en Colombia desde hace varias décadas y su mayor frecuencia se ha demostrado en diferentes estudios 8,16,17,46. En concordancia, los resultados del presente estudio mostraron que una alta proporción (85,7 %) de las muestras caracterizadas pertenecen al subgenotipo F3 del genotipo F. Asimismo, el genotipo A, cuyo origen en África se remonta a 9.600 años, aproximadamente, y el subgenotipo A2, establecido en Europa hace alrededor de 5.000 años 43, han alcanzado una amplia distribución mundial y se pudo identificar en el 14,3 % de las muestras caracterizadas. La circulación de este genotipo en el país había sido reportada en estudios previos 7,16 y el subgenotipo A2 se ha asociado con un mayor riesgo de progresión a la fase crónica de la enfermedad en la población japonesa 47.
Una de las principales limitaciones del presente estudio fue la imposibilidad de obtener información clínica de los pacientes, debido a que las fichas de notificación y las historias clínicas no suministraban la información completa; además, el periodo de estudio fue extenso, lo que impidió completar u obtener los datos clínicos. En este sentido, el estudio se limitó a la descripción de la diversidad de los virus que circularon en el país en el periodo de 2002 a 2014 y de la presencia de mutaciones en el HBV con relevancia epidemiológica.
En la actualidad existen diferentes medicamentos antivirales para el control de la infección por HBV. Sin embargo, la presión selectiva que representan los tratamientos antivirales, la alta tasa de sustitución de nucleótidos del HBV (4,57 x 10-5 sustituciones/nucleótido/año) 48 y el elevado grado de replicación (1012 viriones/día) 49, son factores determinantes que favorecen la aparición de mutaciones de resistencia.
El alto costo de los tratamientos antivirales disponibles y la necesidad de conocer a priori la existencia de mutaciones de resistencia, justifican el estudio de la variabilidad genética de las cepas del HBV que circulan en el país, con el fin de predecir la efectividad de los diferentes tratamientos y contribuir con información útil para la adopción de decisiones.
Teniendo en cuenta que las muestras analizadas en el presente estudio fueron remitidas para el diagnóstico de pacientes con sospecha de hepatitis viral, se puede inferir que estos no habían recibido tratamiento antiviral. Por lo tanto, la identificación de dos mutaciones de resistencia (L180M y M204V) de forma simultánea en el HBV de la muestra proveniente de un paciente de Cundinamarca, sugiere que este adquirió la infección a partir del contacto con un individuo previamente tratado con LdT, 3TC, ETV o emtricitabine (FTC). La mutación I169L en el dominio RT del HBV se identificó en una muestra procedente del departamento del Cesar; aunque es más frecuente encontrar la mutación I169T en dicho dominio, y esta se ha asociado claramente con resistencia a ETV, la presencia de leucina en esta posición podría estar relacionada también con dicha resistencia, y es así como fue interpretada por las herramientas en línea HBV DRI y Geno2Pheno.
Por último, se estima que el surgimiento de variantes de escape en la secuencia que codifica el determinante “a” o en regiones cercanas, tendrá importantes implicaciones en salud pública 23,50. Estas mutaciones están relacionadas principalmente con el escape frente a la neutralización por parte de los anticuerpos generados tras la inmunización con la vacuna, o por infección natural por el HBV, y podrían tener un impacto negativo en el diagnóstico mediante pruebas serológicas basadas en la detección del HBsAg 23,51.
En el análisis de las secuencias del gen S del HBV, se identificó la mutación P120Q en la muestra de un paciente proveniente del departamento del Guaviare, la cual fue reactiva para el HBsAg con el método ELISA. Dicha mutación se ha relacionado con una reacción disminuida en diferentes ensayos serológicos comerciales, como el Vitros ECi HBsAg™ (Ortho-Clinical Diagnostics, Amersham, Buckinghamshire, Reino Unido) y el Auszyme Dynamic™ (Abbott Laboratories, North Chicago, USA) 52,53. Asimismo, se reportó la aparición de esta mutación en un paciente recién nacido de una madre infectada en Corea, a quien se le había administrado de forma simultánea la vacuna y el tratamiento profiláctico con inmunoglobulina 54.
En la región preS/S comúnmente se presentan deleciones 55 y mutaciones puntuales 56, las cuales se han asociado con falla hepática aguda, cirrosis y hepatitis colestásica ‘fibrosante’ 22,57,58. En el presente estudio, dos de las muestras correspondientes al periodo de 2002 a 2003 presentaron una deleción en la región preS1- preS2, cuyas implicaciones bológicas y clínicas se estudiarán y discutirán en estudios posteriores.
El presente estudio permitió corroborar la circulación en Colombia de los genotipos y subgenotipos F3 y A2 del HBV, así como la circulación predominante del subgenotipo F3. Se identificaron las mutaciones L180M y M204V de resistencia a los medicamentos antivirales, cuya presencia simultánea en la secuencia se detectó en la muestra proveniente de un mismo paciente; en la secuencia de otro paciente, se identificó la mutación I169L, la cual también podría conferir resistencia antiviral. En la región codificante del HBsAg, se identificó la mutación P120Q, la cual se ha considerado como variante de escape. Además, se encontró una deleción de 105 nucleótidos que se extendían a la región codificante de preS1 y de preS2, cuyas implicaciones biológicas y clínicas se desconocen.
El presente estudio constituye un aporte a la epidemiologia molecular del HBV en Colombia y a la identificación de mutaciones asociadas con resistencia a los medicamentos antivirales y con mutaciones de escape en las regiones codificantes S y el dominio RT de la polimerasa, lo cual constituye una información valiosa para la adopción de decisiones durante el proceso de elección y seguimiento de la efectividad de los tratamientos antivirales.

