











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
Introducción
El Síndrome de Cohen (OMIM #216550) es una enfermedad genética rara, pocos casos han sido reportados y no se conoce su prevalencia, aunque ha sido descrita en todo el mundo; se sabe que hay más de 100 casos individuales reportados y se estima que entre 500 - 1000 personas han sido diagnosticadas, en casi todos los continentes y con Finlandia aportando más de 35 casos 1.
El mecanismo de herencia es autosómico recesivo, consistiendo en mutaciones en el gen VPS13B. Se caracteriza por obesidad, retraso psicomotor, microcefalia, hipotonía, miopía progresiva, distrofia retiniana, neutropenia intermitente y rasgos faciales particulares 2.
Se presenta el segundo caso reportado en Colombia, confirmado mediante estudio molecular realizado, que consistió en secuenciación completa del gen, mientras que el primero se diagnosticó por hibridación genómica comparativa por microarreglos 3.
Reporte de caso
Adolescente femenina de 14 años, con antecedente de microcefalia desde etapa prenatal. Presentaba retardo del desarrollo psicomotor y a la edad de 7 años en vista de su trastorno cognitivo, microcefalia y malformaciones menores asociadas, fue remitida a valoración por genetista.
Al examen físico durante la primera consulta de genética, se encontraron los siguientes hallazgos: microcefalia, fisuras palpebrales oblicuas dirigidas hacia arriba, pabellones auriculares rotados posteriormente, raíz nasal alargada, filtrum corto, paladar alto, incisivos prominentes, hipoplasia de labios menores, hiperlaxitud, braquidactilia, hipotonía (Figura 1).

Figura 1 Características de la paciente: A - Rasgos faciales y distribución de tejido adiposo a predominio abdominal. B - Manos de la paciente con braquidactilia leve y dedos en forma cónica. C - Fisuras palpebrales oblicuas dirigidas hacia abajo, raíz nasal alargada, filtrum corto e incisivos prominentes. D - Pabellones auriculares rotados posteriormente y raíz nasal alargada.
Con base en lo anterior, se realizaron cariotipo y Estudio de Hibridación Genómica Comparativa array (aCGH) que fueron normales. Quedó en seguimiento clínico desde entonces.
A la edad de 12 años, presentó neutropenia y se diagnosticó obesidad. Se sospechó síndrome de Cohen, por lo que se solicitó secuenciación completa del gen VPS13B que reportó mutación homocigota c.5998_5999del (p.Leu2000Alafs*2), confirmando el diagnóstico.
Discusión y revisión de la literatura
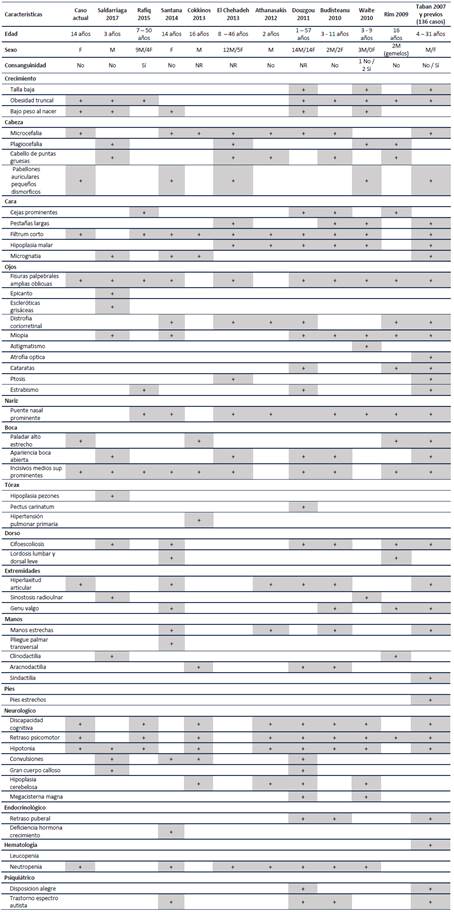
El síndrome de Cohen fue descrito en 1973, con un mecanismo de herencia autosómica recesiva, caracterizado por retraso mental moderado a severo y no regresivo (casi siempre pueden caminar, 20% tienen comunicación verbal), disposición feliz y sociable, microcefalia, hipotonía, actitud corporal ondulada, parpados abiertos inclinados, cejas gruesas y pestañas, surco naso labial corto que deja ver dientes superiores, granulocitopenia, ametropía y distrofia retinocoroidal 4. Se presentan las características fenotípicas reportadas en la literatura comparándolas con el caso presentado, en la Tabla 1. En los datos presentados allí, se observa que no es infrecuente el diagnóstico tardío (en la adultez) y que su ocurrencia pareciera mayor en el sexo masculino, sin un predominio marcado 5-10.
Esta patología se origina por una mutación en el gen vacuolar protein sorting 13B (COH1 ó VPS13B, OMIM 607817) localizado en 8q22 el cual tiene un rol en la separación mediada por vesículas y en el tráfico intracelular de proteínas, lo cual le da participación en gran variedad de tejidos fuera del neuronal, donde además de regiones cerebrales se expresa también en el cerebelo 11,12. Se han identificado más de 213 mutaciones hasta la fecha, según la Human Gene Mutation Database - HGMD 13. La mutación encontrada en este caso solo se ha reportado en una familia 14.
Antenatalmente, sólo se ha descrito que hasta un 50% de los casos presentan hipoactividad y un crecimiento fetal en el límite inferior de la normalidad. Al nacer, la hipotonía puede ocasionar problemas de alimentación, además de un llanto agudo y débil 4,7.
La microcefalia suele desarrollarse durante el primer año, suelen tener un perímetro cefálico normal al nacer 15. En el caso que presentamos es llamativo que está referida la microcefalia desde recién nacida, ocurriendo quizá por la mutación inusual que porta.
La miopía es un sello distintivo, con la característica particular de ser una miopía refractiva y de edad joven, en tres cuartas partes de los casos (lo usual en la miopía juvenil es que sea de origen corneal) 16. Dentro de los problemas oculares, también se presentan ceguera nocturna, campos visuales restringidos, retinitis pigmentaria y alteraciones del iris 16-18. Fue notorio que en el caso presentado no hubo reporte de ametropía.
Los rasgos faciales más notorios del síndrome de Cohen que son el cabello grueso, las cejas prominentes, las fisuras palpebrales oblicuas, el puente nasal prominente, las orejas pequeñas y con algunos rasgos dismórficos, el filtrum corto, la apariencia de boca abierta con incisivos prominentes y la micrognatia, pueden no ser tan notorios en preescolares, pero se pueden asociar al diagnóstico si se tiene presente buscarlos en niños con miopía, trastornos conductuales y/o neutropenia 6. En el caso expuesto no se pudo establecer la asociación sino hasta la aparición de la alteración hematológica.
A nivel endocrinológico, se pueden presentar talla baja y pubertad retrasada, asociadas a la obesidad 15. La patología cardiaca también es de baja frecuencia, pudiendo hallarse hipertensión pulmonar y prolapso mitral, que se pueden comportar como patologías silentes por un largo tiempo 19.
La neutropenia es leve a moderada, no es cíclica y no es fatal. Suelen encontrarse infecciones recurrentes y úlceras aftosas asociadas. Los recuentos van entre 500 - 1200/mm3. Puede presentarse sin leucopenia. En algunos casos, podría asociar déficit de proteína C y S 2,4.
La hipotonía mejora con el tiempo (sin causa clara), pero se asocia hipermovilidad, cifosis, escoliosis y pie plano 2,20.
Pocos pacientes se han encontrado con características autistas 7, siendo más común que tengan una actitud amigable y una personalidad interactiva 2,20.
En cuanto al manejo de sus patologías, debe ofrecerse corrección con lentes y/o cirugía para la miopía, terapia física, tratamiento de las infecciones, considerar si requieren factores estimulantes de colonias de granulocitos, manejo endocrinológico, seguimiento por neurología, asesoría genética y apoyo psicológico 2.
En conclusión, a pesar de ser un síndrome poco común, con importante variabilidad fenotípica, debe sospecharse con base en los criterios clínicos y en las patologías asociadas, que pueden ser de expresión clínica lenta. Es una causa de obesidad genética, alteraciones hematológicas (fundamentalmente neutropenia), ametropías severas (principalmente miopía) y autismo, que pueden ser las claves en el diagnóstico, como en el caso presentado. Confirmando el diagnóstico se puede dar una adecuada asesoría genética, así como mejorar la calidad de vida de estos pacientes.

