











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroduction
Phytoplasmas are bacteria of the class Mollicutes that lack cell walls and are sensitive to tetracycline. It has been stated that they inhabit the phloem cells of plants or the intestine, haemolymph, salivary glands and other internal organs of some hemipteran insects of the families Cicadellidae, Fulgoromorpha, and Psyllidae (Lee et al., 2000; Weintraub and Beanland, 2006; Hogenhout et al., 2008; Bertaccini and Duduk, 2009). Phytoplasmas are difficult to cultivate (Contaldo et al., 2016), so their detection, identification and classification are mainly based on the amplification of the 16SrRNA gene (Gundersen et al., 1996) and further analysis by restriction fragment length polymorphism (RFLP) with 17 restriction enzymes that discriminate them into ribosomal groups. To date, 34 16Sr groups are recognized for phytoplasmas (Pérez-López et al., 2016). The genus 'Candidatus Phytoplasma' was adopted for incompletely described, non-cultivable phytoplasmas. A new species within Candidatus is created when the sequence of the 16SrRNA gene contains less than 97.5% of nucleotide identity compared to previously described phytoplasma species (IRPCM, 2004).
The presence of phytoplasmas is associated with diseases in crops, ornamentals, fruit and woody plants belonging to different botanical families (Lee et al., 2000; Bertaccini et al., 2014). In Colombia, phytoplasmas from the 16SrIII group were reported in Coffea arabica, Cordia alliodora, Physalis peruviana and Solanum quitoense (Galvis et al., 2007; Mejía et al., 2014); 16SrV and 16SrXII in Solanum tuberosum (Mejía et al., 2011; Varela and Franco-Lara, 2017); 16SrIII-L in Manihot esculenta (Alvarez et al., 2009); 16SrIX in Catharanthus roseus (Duduk et al., 2008a), and 16SrI in Elaeis guineensis, Zea mays and Musa acuminata (Duduk et al, 2008b; Alvarez et al, 2014; Aliaga et al, 2018). In Bogota, phytoplasmas of the groups 16SrI and 16SrVII are widely distributed in urban trees such as Acacia melan-oxylon, Croton spp., Eugenia neomyrtifolia, Fraxinus uhdei, Liquidambar styraciflua, Magnolia grandiflora, Pittosporum undulatum, Populus nigra, and Quercus humboldtii (Perilla-Henao et al., 2012; Perilla-Henao and Franco-Lara, 2013; Franco-Lara and Perilla-Henao, 2014; Franco-Lara et al., 2017). Phytoplasma diseases in strawberry Fragaria x ananassa have been reported in Australia (Padovan et al., 1998; Padovan et al., 2000), the United States (Harrison et al., 1997; Jomantiene et al., 1999), France (Danet et al., 2003), Italy (Bertaccini et al., 1997), and Lithuania (Valiunas et al., 2006). In Colombia, phytoplasma groups 16SrI and 16SrVII have been reported in strawberry varieties Camino Real and Ventana, with symptoms of flower phyllody, virescence of achenes, double sepals and aborted fruits (Perilla-Henao and Franco-Lara, 2014).
Amplicephalus funzaensis Linnavuori 1968 (Cicadellidae: Deltocephalinae) and Exitianus atratus Linnavuori 1959 (Cicadellidae: Deltocephalinae) are known vectors of phytoplasma groups 16SrI and 16SrVII in Bogota (Perilla-Henao et al., 2016). The objective of this study was to determine if A. funzaensis can transmit phytoplasmas to Fragaria x ananassa, and to generate a DNA barcode for this insect species.
Materials and methods
The transmission assays were carried out at the campus of the Universidad Militar Nueva Granada (UMNG), in Cajica, Cundinamarca (4°55'0" N, 74°1'30" W), at an altitude of2558 m a.s.l., with average annual temperature of 13.9°C and average annual relative humidity of 87.9% (information obtained in 2016 from the meteorological station at Nueva Granada Campus, when the assays were carried out).
Entomological material
Insects were collected from pastures represented mainly by Cenchrus clandestinus using an entomological net and a mouth aspirator. Samplings were performed at the campus UMNG or at the Universidad Nacional de Colombia (UN) campus in Bogota, Colombia (4°38'18.88" N, 74°5'2.76" W). The insects were stored in 90% (v/v) ethanol. Taxonomic identification was based on morphological characters (Linnavuori, 1959; Dietrich, 2005; Silva-Castaño et al., 2019). Before performing the transmission experiments, a natural population of A. funzaensis that lived on the grass C. clandestinus at the campus UMNG in Cajica was tested for the presence of phytoplasmas. In July 2015, 20 individuals were analyzed by nested PCR to test for phytoplasmas. The A. funzaensis used for the transmission assays were captured in April 2016 at the campus UMNG from the population in which the natural infection by phytoplasmas had been recorded. In February and March 2016, another group of twelve A. funzaensis belonging to two different populations, UMNG campus and UN campus were used to generate a DNA barcode for this species.
Transmission assays
One hundred adult A. funzaensis were collected and maintained for 5 min at 4°C to reduce their mobility and separate females and males, before placing them on strawberry F. x ananassa variety Monterrey of approximately 6 months old, grown in an aquaponic culture. The plants were propagated by division of the crown and the new plants were potted individually in 7 kg pots using soil and rice husks (2:1) as substrate. Before the experiment, the plants were kept for three months in insect free cages and were watered every 3 d by drip irrigation and fertilized once a month with Hoagland and Arnon (1950) nutrient solution. To confirm that all the plants were free of phytoplasmas before the transmission assays, nested PCR tests were performed, as described below. Treatment 1 consisted of two replicas with five plants each, and treatment 2 (control) consisted of one replica with five plants that were not exposed to A. funzaensis hervibory. Plants of both treatments were kept in independent insect free cages. For treatment 1, each plant was infested with ten male and female adults of A. funzaensis captured at the campus UMNG in Cajica, from the population in which phytoplasmas had been previously detected. To guarantee that the insects were in contact with the leaves, two 10 cm X 10 cm veil bags containing five insects each were tied to different leaves of each plant.
When the insects died, 1 week later, they were retrieved and stored at -20°C for molecular testing. Two months post-herbivory, leaves of all the plants of treatments 1 and 2 were sampled and tested for phytoplasmas. The plants were kept for 7 months in observation inside cages and monthly observations were made to register the appearance of symptoms. During these months, watering and fertilization were performed as described before.
DNA extraction
Insect DNA extractions were performed following the methodology described by Hung et al. (2004). One A. funzaensis individual was ground with liquid nitrogen and 30 µl of lysis buffer (0.1 M Tris-HCl pH 8.0, 0.05 M EDTA, 0.01% (v/v) N-Lauroylsarcosine). After grinding, 970µl of the same buffer were added. The tissue was treated with 1% (w/v) sodium dodecyl sulphate and 0.08 mg ml-1 proteinase K, and incubated at 55°C overnight. The next day, 500µl of 1:1 equilibrated pH 8.0 phenol and chloroform were added and the sample was centrifuged at 10,000 g for 10 min. The aqueous phase was removed and the nucleic acids precipitated with 1 ml of absolute ethanol for 15 min at -20°C, and then samples were centrifuged at 10,000 g for 25 min. The pellet was washed in 1 ml of 70% (v/v) ethanol. The supernatant was eliminated and the ethanol left to evaporate. Finally, the DNA was resuspended in 50µl TE buffer (10 mM Tris-HCL, 1 mM EDTA pH 8.0) and stored at -20°C. The DNA concentration of the samples was quantified in a Qubit (Invitrogen®) using the Qubit Assay kit.
The plant DNA extractions were performed using a protocol by Dr. A. Bertaccini (personal communication) and adapted in our laboratory. Extractions started from 0.33 g of leaf midribs. The midribs were ground in liquid nitrogen and extraction buffer I (92 mM K2HPO4 x 3H2O, 30 mM KH2PO4, 10% sucrose (w/v), 5% (w/v) PVP-10 and L-ascorbic acid (pH 7.6)). The samples were filtered through sterile cloths, centrifuged at 6500 g for 10 min and resuspended in 1.3 ml of extraction buffer II (100 mM Tris-HCL, 100 mM EDTA, 250 mM NaCl) with 25µl of proteinase K (5 mg/ ml) and 145µl of N-Lauroylsarcosine 10% (w/v). Samples were incubated at 55°C for 2 h. The lysate was centrifuged for 10 min at 8,000 g and the pellet discarded. The nucleic acids were precipitated with 0.6 volumes of isopropanol and incubated at 4°C overnight. Next day, the extracts were centrifuged at 8,000 g for 30 min and the pellet resuspended in 990 nl of TE buffer (10 mM Tris-HCl, 1 mM EDTA [pH 8.0], 24.75 ml of 20% (w/v) sodium lauryl sulphate and 19.8µl of proteinase K (5 µmg ml-1)), and incubated at 37°C for 1 h. Later, 175µl of 5 M NaCl and 140µl (10% (w/v) cetyl trimethyl ammonium bromide/0.7M NaCl) were added and the lysate incubated at 65°C for 10 min. Two consecutive extractions were performed, the first with an equal volume of chloroform / isoamyl alcohol (24:1) and the second with an equal volume of chloroform. In both cases the phases were separated by centrifugation at 8,000 g for 10 min and the organic phase discarded. The supernatant was precipitated with 0.6 volumes of isopropanol at 4°C overnight. Finally, the nucleic acids were pelleted by centrifugation at 11,000 g for 20 min and the pellet washed with ethanol 70%. The pellet was resuspended in 50 of TE buffer and the DNA was stored at -20°C. Later, the DNA was purified with the PowerClean® Pro DNA Clean-Up kit (Mo Bio Laboratories, Inc.) to eliminate other impurities.
Amplification of the COI gene of A. funzaensis and sequence analysis
The amplification of a region of the COI mtDNA gene was performed with the universal insect primers LCO1490/ HCO2198 (Folmer et al, 1994). The PCR reactions were conducted at a final volume of 25 ^l, with 0.05 U of Taq polymerase (Bioline®), 1X reaction buffer, 2.5 mM MgCl2, 0.3 mM dNTPs, 0.3 of each primer and 50 ng of DNA diluted in deionized water. The thermic profile consisted on a denaturation for 10 min at 95°C, followed by 35 cycles of denaturation for 1 min at 95°C, annealing for 45 s at 40°C, extension for 45 s at 72°C and, a final extension period for 10 min at 72°C. The amplicons were purified with the UltraClean® PCR Clean-up Kit (Mo Bio Laboratories, Inc.) and were bi-directionally sequenced in Macrogen, Korea. The sequences were edited with the Geneious R9 software (Biomatters Ltd.) and each individual consensus sequences were built by superposition of the forward and reverse sequences. The consensus sequences were compared using BLASTN with the GenBank database. Additionally, nine A. funzaensis sequences obtained in this research, and other Cicadellidae downloaded from the BOLD database (http://www.barcodinglife.org) were used to construct a distance tree by the neighbor-joining method, with 1000 bootstrap, based on the Kimura 2 parameter (K2P) model, using the Mega v.6.0 software (Tamura et al., 2013). A sequence of Psylla galeaformis (Hemiptera: Psyllidae) was used as out-group. The Barcode Gap analysis of the COI gene sequences was performed by comparison of the intraspecific versus the interspecific sequences, obtained from the distance matrix generated in MEGA v.6.0, according to the K2P model.
Detection of phytoplasmas using PCR
The presence of phytoplasmas was tested by PCR in F. x ananassa and A. funzaensis DNA extracts. To verify that PCR inhibitors were not present in the plant DNA extracts, amplification of rps16 chloroplast intron was performed, before phytoplasma detection (Oxelman et al, 1997). The PCR reactions were conducted in a final volume of 15 µl, with 0.05 U of Biolase (Bioline), 1X reaction buffer, 1.5 mM MgCl2, 0.2 mM dNTPs and 0.2 of primers. Dilutions 1:50 and 1:100 were prepared from the plant DNA extracts in deionized water and 2µl were used as template. Deionized water was used as negative control in all the PCR reactions. The thermic profile was: initial denaturation for 5 min at 95°C, followed by 35 cycles of denaturation for 1 min at 94°C, annealing for 1 min at 53°C, extension for 2 min at 72°C, and a final extension period for 10 min at 72°C.
The PCR reactions for phytoplasmas were made as explained before. For F. x ananassa, 1:20, 1:50 and 1:100 dilutions of the DNA extracts were tested. For the insects, 1:20 dilutions of DNA extracts were analyzed. The PCR with universal primers P1A/P7A (Lee et al., 2004) produced amplicons of ~1800 bp, which were diluted 1:20 and used in nested PCRs with the internal primers R16mF2/ R16mR1 (Gundersen and Lee, 1996) to obtain ~1430 bp amplicons. Frequently, when low intensity amplicon bands were observed in nested PCRs, the resulting amplicons were amplified again using internal primers R16F2n/ R16R2 (Gundersen and Lee, 1996) using 1:20 dilutions of the amplicon as template. Bands with enough DNA were further analyzed by sequencing or RFLP analyses. In these reactions, the positive control corresponded to fragments of the region 16S-23S of the phytoplasmas Maize bushy stunt - MBS (16SrI-B group) and ash yellows ASHY (16SrVII-A group), previously cloned and maintained in an Escherichia coli strain. In the F. x ananassa plants and in the A. funzaensis, additional nested PCRs tests were performed with different primer combinations such as P1A/P7A (Lee et al, 2004) followed by R16F2n/R16R2, and R16mF2/R16mR1 in primary reactions followed by R16F2n/R16R2. The plant DNA extracts were analyzed in 1:50 and 1:100 dilutions and the insect DNA extracts in 1:10 dilutions. The thermic profile was as described before, except for annealing temperatures that depended on the primer pairs used: P1A/P7A, 55°C; R16mF2/R16mR1, 54°C and R16F2n/R16R2, 54°C. The amplicons were separated using standard electrophoresis in 1% agarose gels in TBE buffer 1X (89 mM Tris base, 89 mM boric acid, 2 mM EDTA), stained with ethidium bromide and visualized under UV light.
To determine phytoplasma ribosomal group affiliation, nested PCR amplifications were performed with the R16F2n/R16R2 primers at a final volume of 30µl. This volume was divided into four, and each aliquot digested with the restriction endonucleases Rsal, Msel, fihal or AluI (New Englad BioLabs). Digestions were conducted at a final volume of 20µl, with 0.05 U of enzyme, 1X buffer and 6.5µl of the PCR product obtained by nested amplification. The reactions were incubated at 37°C overnight. The restriction products were separated using agarose gel electrophoresis at 3% in TBE buffer. Additionally, the selected amplicons obtained with the primers R16F2n/R16R2 were purified with the UltraClean® PCR Clean-up Kit (Mo Bio Laboratories, Inc.) and sequenced bidirectionally in Macrogen, Korea. The sequence analysis was performed as explained before.
Results
Entomological material
Only specimens of A. funzaensis, confirmed by morphological and molecular methods, were included in this study (Fig. 1).

Transmission assays
At the time of this experiment it was not possible to raise A. funzaensis under laboratory conditions, so the transmission experiment was performed with insects from a naturally infected population. In a previous work, a population of A. funzaensis infected with 16SrI and 16SrVII phytoplasmas had been found, but before performing the experiment it was tested again for the presence of phytoplasmas. Twelve adults of twenty A. funzaensis from that population were found positive for phytoplasmas by nested PCR, but no further RFLP or sequencing analysis were performed because the quantity of the obtained DNA was too small.
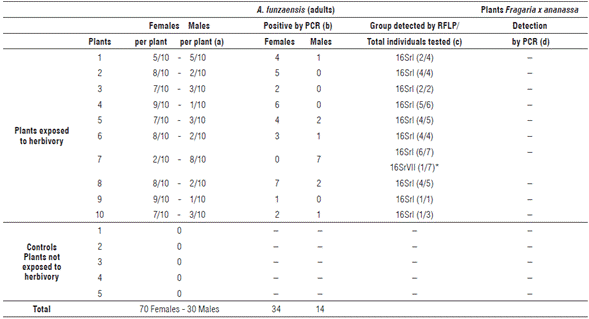
For the transmission experiment, a different set of 100 insects were allowed to feed in 10 phytoplasma-free plants, placing 10 insects per plant. DNA extraction is a destructive method, so the detection of the phytoplasmas in these insects was performed after they were allowed to feed on the test plants. After two months post-herbivory, all the F. x ananassa plants were negative for phytoplasmas, regardless of the PCR primer combination used, as were the non-exposed negative control plants. In all the cases, the positive PCR controls generated bands of the expected size and the negatives did not amplify (Figs. 2 A-C). No symptoms associated with phytoplasmas in leaves, flowers, or fruits were observed after 7 months of evaluation (Tab. 1).
TABLE 1 Results of transmission assays of phytoplasmas by Amplicephalus funzaensis to Fragaria x ananassa in semi-controlled conditions.
(a) Number of females and males placed on each tested plant, over total number of insects. (b) Number of females and males positive for phytoplasmas by nested PCR.
(c) Number of positive insects for 16SrI or 16SrVII groups, determined by RFLP with restriction enzymes fisaI, MseI, HhaI and AluI, over the total of tested insects.
(d) Detection of phytoplasmas in F. x ananassa by nested PCR and double nested PCR 2 months post-inoculation.
(-) Negative for phytoplasmas.
(*) Samples with mixed infections containing 16SrI and 16SrVII.
The control plants were also negative for the presence of phytoplasmas in tests conducted simultaneously with plants exposed to insects. In all cases, the positive controls generated bands of the expected size and the negatives did not amplify (Figs. 2 A-C).

FIGURE 2 Nested and double nested PCR of Fragaria x ananassa samples exposed to Amplicephalus funzaensis (Treatment 1) and control plants (Treatment 2). (A) Double nested PCR with primers P1A/P7A, R16mF2/R16mR1 followed by R16F2n/R16R2. (B) Nested PCR with primers P1A/ P7A and R16F2n/R16R2. (C) Nested PCR with primers R16mF2/R16mR1 and R16F2n/R16R2. In all the cases, only the result of the last PCR is shown. MBS: Positive control group 16SrI-B. ASHY: Positive control group 16SrVII-A. Negative control: water. II: Molecular weight marker Hyper-ladder 50 pb (Bioline®).
Of 100 A. funzaensis used in the transmission assays, 22 individuals resulted positive for phytoplasmas by nested PCR with primers P1A/P7A followed by R16F2n/R16R2 and 36 individuals were positive with primers R16mF2/ R16mR1 and R16F2n/R16R2. Taking into account nested PCR tests with both sets of primers, 48 of 100 individuals that were used in the transmission tests were positive in PCR using primers for phytoplasmas (Fig. 3, Tab.1).
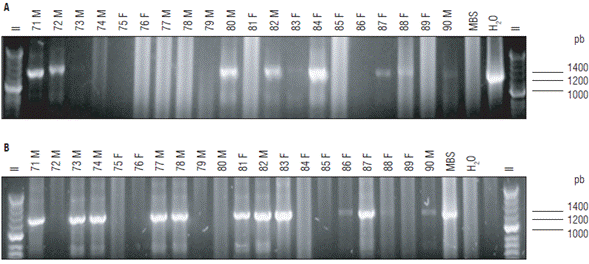
FIGURE 3 Detection of phytoplasmas by nested PCR in the Amplicephalus funzaensis used in transmission assays. (A) Nested PCR with primers P1A/P7A followed by R16F2n/R16R2. (B) Nested PCR with R16mF2/R16mR1 followed by R16F2n/R16R2. Only the result of the nested PCR is shown. The number corresponds to the insect code, F: females and M: males. MBS: Positive control group 16SrI-B. Negative control: water. II: Molecular weight marker Hyperladder 50 pb (Bioline®).
Of the 48 positive insects, 41 were analyzed by RFLP. Of these, 34 were infected with phytoplasmas: 33 were infected with 16SrI group and one simultaneously with 16SrI and 16SrVII groups (Tab. 1). In the others, the RFLP patterns obtained were not consistent with any phytoplasma group profile, suggesting non-specific amplifications (Fig. 4).
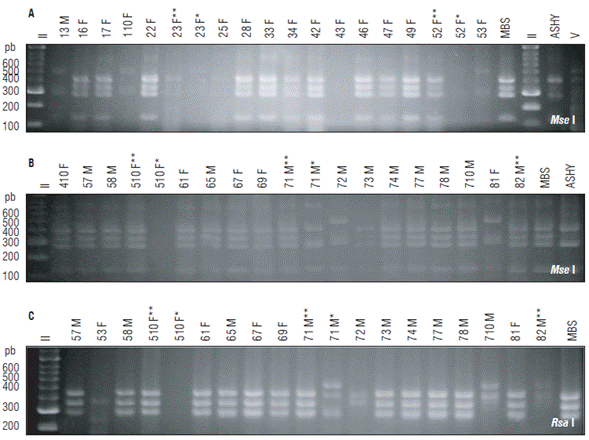
FIGURE 4 Detection of phytoplasmas in A. funzaensis used in the transmission assays. (A-B) RFLP with MseI, (C) RFLPs with flsaI, of amplicons obtained by nested PCR with primers P1A/P7A, R16mF2/R16mR1 and R16F2n/R16R2. *Nested PCRs with P1A/P7A followed by R16F2n/R16R2. **Nested PCR with R16mF2/R16mR1 followed by R16F2n/R16R2. The number corresponds to the insect code, F: Females and M: Males. MBS: Positive control group 16SrI-B. ASHY: Positive control group 16SrVII-A. Negative control: Water. II: Molecular weight marker Hyperladder 50 bp (Bioline). V: Molecular weight marker Hyperladder 25 bp (Bioline®).
DNA barcoded for A. funzaensis
Amplicons of the mitochondrial DNA COI gene were obtained from 12 individuals, six collected at the UMNG and six at the UN. Consensus sequences of lengths between 667 and 706 bp were obtained from nine individuals (Fig. 5A); the other sequences were illegible. The edited sequences were uploaded in the BOLD database (http://v3.boldsystems.org/) (Ratnasingham and Hebert, 2007). The sequences of all the A. funzaensis collected in Bogota at the UN showed a conserved haplotype (Sequences ID: COLCI053-18, COLCI054-18, COLCI055-18, COLCI056-18, and COLCI057-18), but the ones collected at the UMNG in Cajica represented three haplotypes. Haplotype 1 was identical to the Bogota sequence (Sequence ID: COL-CI049-18); two sequences called haplotype 2 (Sequences ID: COLCI051-18 and COLCI052-18) presented substitutions T by C at positions 183, A by G in 453 and T by C in 517. In 99%. Sequences for A. funzaensis were not found in the haplotype 3, there was a substitution of G by A in position BOLD or GenBank databases, therefore, Deltocephalinae 342 (Sequence ID: COLCI050-18). The sequence similarity sequences showing similarities between 86% and 87% were of the COI sequences obtained for A. funzaensis was over used for comparison.

FIGURE 5 (A) Amplification of the COI gene of Amplicephalus funzaensis by PCR. Negative control: water. II: Molecular weight marker Hyperladder 50 pb (Bioline). (B) Distance tree constructed by neighbor-joining (NJ) based on the Kimura 2 parameter model (K2P) with 1000 bootstrap, using 25 COI sequences of the genus Deltocephalini, which includes Amplicephalus. The out group was a sequence of Psylla galeaformis. Insects collected at UMNG are shown with triangles, and at UN with circles.
The nine A. funzaensis sequences obtained in this work formed a distinct cluster from other sequences of the Amplicephalus genus obtained from BOLD and GenBank. All the sequences clustered by taxa, showing that they had enough differences to separate them as distinct groups (Fig. 5B). The average intraspecific genetic divergence varied between 0.0000 and 0.0245 (mean 0.0048) and the interspecific divergence between 0.1003 and 0.2341 (mean 0.1501). The most divergent species were P. flavicosta compared to A. simplarius (0.2341), A. curtisii compared to A. littoralis (0.2233) and P. flavicosta compared to A. littoralis (0.2233). The less divergent species were A. littoralis and A. simplarius (0.1003). The A. funzaensis sequences were close to D. balli (0.1499) and A. curtisii (0.1739), and showed the largest divergence with A. simplarius (0.2004). The barcode gap analysis showed that for the eight species analyzed, the sequence variability at the intraspecific level was lower than the variability at the interspecific level.
Discussion
A very important aspect of the epidemiology of the diseases associated with phytoplasmas is the identification of their insect vectors. To do this, the insects nearby the symptomatic plants are collected and their ability to transmit phytoplasmas to healthy host plants is tested (Weintraub and Beanland, 2006). The presence of phytoplasmas in an insect does not guarantee its vector ability; therefore, transmission tests are mandatory (Purcell et al., 1981; Vega et al., 1993). In this work, tests were performed with A. funzaensis because it is a known vector of phytoplasma, and it is ubiquitously distributed in the C. clandestinus grasslands of the Bogota plateau that frequently surrounds F. x ananassa crops (Perilla-Henao et al., 2016; Silva-Castaño et al., 2019).
No evidence was observed of phytoplasma transmission by A. funzaensis to F. x ananassa. The plants exposed to infected A. funzaensis were negative by PCR 2 months post-herbivory and no symptoms were observed for seven months, although the observations performed during the transmission experiment suggested that the insects fed from the test plants and that 36 of the 100 insects used in the assay were infected with phytoplasmas of 16SrI or 16SrVII groups. Several explanations for these results are possible. For instance, that the F. x ananassa variety Monterrey plants are not susceptible to phytoplasmas. However, more tests would be necessary to confirm this possibility taking into account that phytoplasma infections in strawberry are widely reported (Padovan et al., 1998; Jomantiene et al., 1999; Padovan et al., 2000; Danet et al., 2003; Valiunas et al., 2006). Furthermore, phytoplasmas of groups 16SrI and 16SrVII have been detected in strawberries of Camino Real and Ventana in Cundinamarca before (Perilla-Henao and Franco-Lara, 2014). Taking into account that the PCR tests were performed 2 months post-herbivory, it is also possible that the plants at that moment were infected but the phytoplasma title was under the PCR detection level. It would have been desirable to test for the presence of phytoplasmas by PCR in the following period, but the plant tissue collected in the subsequent months was lost due to refrigeration problems. Symptoms were not observed in the next seven months in the plants exposed to insects. Furthermore, it has been known that in different plant hosts, several groups of phytoplasmas can accumulate either in the source tissues, such as in Euphorbia pulcherrima and Cathatantus roseus with levels of accumulation that can vary one order of magnitude being more concentrated in C. roseus than in E. pulcherrima (Christensen et al., 2004), or in the case of Australian Papaya Dieback Phytoplasma which accumulated in young leaves but not in fruits (Sid-dique et al., 1998), making it difficult to select the sampling tissue in an unknown host.
When this research was carried out, a rearing for A. funzaensis was unavailable. In consequence, many variables that influence the transmission efficiency were not controlled. The transmission of phytoplasmas by their insect vectors is of the circulative, propagative type, and occurs in three phases (Weintraub and Beanland, 2006; Perilla-Henao and Casteel, 2016). The acquisition access period (APP) is the feeding time necessary for the insect to acquire phytoplasmas, and the latency period (LP) is the period elapsed between the acquisition and the moment in which the insect is able to transmit phytoplasmas. During LP, the phytoplasmas replicate inside the insect and only when the salivary glands become infected, transmission occurs (Weintraub and Beanland, 2006). The latency period can take as long as 3 weeks (Webb et al., 1999). The inoculation access period (IAP) is the time during which the insects are infective, which in this case is all their lives (Alma et al., 2019). It is known that insects that test positive for phytoplasmas by nested are not necessary vectors (Purcell et al., 1981; Vega et al., 1993); to be able to transmit, phytoplasmas must infect the salivary glands of the insect (Weintraub and Beanland, 2006; Alma et al., 2019). In this case, the DNA extractions performed on the A. funzaensis individuals were performed from the whole body and not only from the head, so it is impossible to know if the salivary glands were infected. The efficiency of transmission is also affected by the insect species. Euscelidius variegatus (Hemiptera: Cicadellidae) is considered to be an inefficient insect vector of the Crisantemum Yellows (CY) phytoplasma (group 16SrIB), in comparison to Macrosteles quadripunctulatus (Hemiptera: Cicadellidae). It has shown that in E. variegatus about 75% of the individuals get their salivary glands infected with CY in experimental conditions in comparison to M. quadripunctulatus in which 100% of the individuals get infected salivary glands. The transmission efficiency is attributed to the fact that the intestine and salivary gland membranes of E. variagatus act as semi-selective barriers to the pass of phytoplasmas, making them less efficient in transmission (Galetto et al., 2009). It was later demonstrated that the CY Amp protein (major antigenic membrane) plays a role in the recognition between the phyotplasm and insect membranes in the early stages of the vector infection (Rashidi et al., 2015). On the other hand, Palermo et al. (2001) showed that in infective individuals of Macrosteles quadripunctulatus (Hemiptera: Cicadellidae), a very efficient vector of CY, the infected insects have periods in which they were unable to transmit, but a few days later they recovered this ability (Palermo et al., 2001). In this test, the infected A. funzaensis were collected from an infected grass patch, but the AAP and LP were not considered. Other variables not considered in this research were the number of insects infected at the beginning of the experiment (Galetto et al., 2009), the host plant from which the phytoplasmas were collected (Palermo et al., 2001), the plant load (Galetto et al., 2014), and the control of temperature which could affect the transmission efficiency (Murral et al., 1996; Maggi et al., 2014). Recommendations for future experiments include a period of pre-adaptation feeding for the insects before the transmission tests, and longer observation periods and using nymphs instead of adults, as it is generally considered that they are more efficient vectors. Nymphs and adults can be infected with phytoplasmas but their transmission efficiency varies, even if they feed on the same part of the plant if nymphs are exposed earlier in their lives to phytoplasmas (Weintraub, 2007; Alma et al., 2019).
The potential transmission ability of insect vectors can vary according to their feeding habits on different plant species, the phytoplasma groups that infect the host plants and the susceptibility of the insect to the colonization of the phytoplasmas, in addition to the intimate molecular relationships that occur between insects and phytoplasmas (Palermo et al., 2001; D'Amelio et al., 2007; Galetto et al., 2009; Bosco and D'Amelio, 2010). There is little information about the interactions between phytoplasmas and different AP and LP which compete for tissues and organs (D'Amelio et al., 2007; Bosco and D'Amelio, 2010; Perilla-Henao and Casteel, 2016), as would be the case of the 16SrI and 16SrVII phytoplasmas.
In this experiment, the A. funzaensis was collected from a natural population of infected insects growing in grass C. clandestinus, a monocot C4 plant, which was placed on strawberry, a dicot C3 plant. It is, therefore, possible that the different diet and nutritional content of the plants, in addition to the stress associated with the experimental conditions found in caged plants inside a greenhouse, may have affected their survival or transmission efficiency.
In total, 120 individuals of A. funzaensis were individually tested for the presence of phytoplasmas, including the insects evaluated to test the population used in the transmission experiments, and the insects were allowed to feed on the test plants. Of these individuals, 38% carried phytoplasmas. Of the 100 insects used for the transmission essays, 33 carried phytoplasmas of the 16SrI group and one carried a mix of 16SrI and 16SrVII groups. The existence of A. funzaensis populations in which large percentages of insects are infected with phytoplasmas make them a potential risk for dispersion of phytoplasmas in the Bogota plateau.
The groups 16SrI and 16SrVII have been reported as the most common phytoplasma groups in the Bogota plateau infecting trees (Perilla-Henao et al., 2012; Perilla-Henao and Franco-Lara, 2013; Franco-Lara and Perilla-Henao, 2014; Franco-Lara et al., 2017). Furthermore, 17% of the positive samples by nested PCR produced RFLP patterns that were inconsistent with phytoplasmas, confirming that not specific amplification of DNA can occur with the phytoplasma primers used, as has been reported before (Abeysinghe et al., 2016).
This is the first report of a COI DNA barcode sequence for A. funzaensis. This sequence allows the separation of this species from other members of the same genus such as A. litoralis, A. osborni and A. simplarius. The two A. funzaensis populations studied were separated by a geographical distance of 35 km. The DNA barcode was represented by a major haplotype shared between the two populations and two additional haplotypes that had a few nucleotide substitutions (average genetic distance of 0.0048). This low genetic variability suggests that A. funzaensis in the Bogota plateau is a meta-population thriving due to the efficient use of C. clandestinus as a host plant, a non-native grass that dominates the landscape. More information is needed to characterize the genetic variability of this species in the zone; in addition, possible differences between its symbionts could enhance its biological efficiency improving its nutritional and reproductive status (Takiya et al., 2006). Among the symbionts, phytoplasmas are known to cause differential effects on the fitness of the insect vectors, in some cases improving their general fitness (Beanland et al., 2000), or on the contrary negatively affecting their life span, fecundity, and progeny size (Bressan et al., 2005).
This work does not provide proof of the ability of A. funzaensis to transmit phytoplasmas to F. x ananassa and more tests are needed. However, the fact that a high number of insects are infected with three groups of phytoplasmas highlights the need for epidemiological surveillance of crops and plants of the urban landscape of the Bogota plateau.

