




![Synthesis and Spectroscopic Characterization of the Products Derived from the Reaction of [Ru3(CO)12] with the Chloroaryliderhodanine Ligand](/img/en/next.gif)





Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista de Ciencias
Print version ISSN 0121-1935
rev. cienc. vol.18 no.1 Cali Jan./June 2014
Determining the Relation between the Proportion of the Amino Group and the Degree of Deacetylation of Chitosan
Determinación de la relación entre el porcentaje del grupo amino y el grado de desacetilación del quitosano
Diana Marcela Escobar Sierra
Programa de Bioingeniería, Grupo de investigación en Biomateriales, Universidad de Antioquia, Medellín - Colombia
E-mail: descobar.udea@gmail.com
Alex Mauricio Castro Ramírez
Bioingeniero, Universidad de Antioquia, Medellín - Colombia
E-mail: mauriciocastro9007@gmail.com
Natalia Andrea Vergara Castrillón
Bioingeniera, Universidad de Antioquia, Medellín - Colombia
E-mail: navefuture@yahoo.com
Received: September 23, 2013
Accepted: November 20, 2013
Abstract
Several methods such as elemental analysis or infrared spectroscopy technique (IR) have been already developed to determine the degree of N-acetylation, for chitin and chitosan. This paper proposed the potentiometric technique to determine the relationship between the amino group proportion and the deacetylation degree of chitosan, using two chitosan samples obtained from different sources: one, by previously established protocols, and a commercial chitosan. The polymers tested were characterized by X-ray Diffraction and Fourier Transform Infrared Spectroscopy to verify the purity of the polymers. The proposed technique allowed us to demonstrate that the molecular weight of the synthesized chitosan are within the range reported for the biopolymer with amino group proportions similar to those obtained for samples of commercial chitosan.
Keywords: deacetylation, potentiometry, chitosan, viscosimetry.
Resumen
Varios métodos han sido desarrollados para determinar el grado de N-acetilación de la quitina y el quitosano, como el análisis elemental, o la espectroscopía de infrarrojo (IR). En este trabajo se propuso emplear el método potenciométrico para determinar la relación entre el porcentaje del grupo amino y el grado de desacetilación del quitosano, para lo cual se emplearon dos muestras de quitosano obtenido de diferentes fuentes (utilizando protocolos establecidos previamente) y un Espectroscopía de Infrarrojo con Transformada de Fourier para corroborar la pureza de los polímeros. La técnica propuesta permitió demostrar que el peso molecular de los quitosanos sintetizados se encuentra dentro de los márgenes reportados para dicho biopolímero, con una proporción del grupo amino muy similar a la obtenida para muestras de quitosano comercial.
Palabras clave: desacetilación, potenciometría, quitosano, viscosimetría.
1. Introducción
El quitosano es un polisacárido de gran abundancia en la naturaleza, que se obtiene como derivado desacetilado de la quitina, que está presente en hongos, algas, insectos y crisálidas del gusano de seda (Paulino et al. , 2006). También se ha encontrado en mayores cantidades en los calamares (Chandumpai et al. 2004), en el exoesqueleto de crustáceos como los cangrejos (Yamaguchi et al., 2003; Vongchan et al. 2003) y langostas (Fernández et al., 2004).
Químicamente, la quitina y el quitosano son poliglucosaminas que son distinguidas únicamente por el grado de la acetilación de los grupos amino, puesto que la quitina tiene generalmente un grado de acetilación entre 70-95% que corresponde a un contenido de acetilo de un 15-20,7%, mientras que el quitosano tiene un grado de N-acetilación entre 15-25% que corresponde a 3,2-5,3% del contenido de acetilo. Siendo el grado de N-acetilación el parámetro más importante que determina sus características funcionales y fisiológicas (Hernández et al., 2009), que lo hace apto para aplicarlo en variados campos como el biotecnológico (Hirano, 1999), biomédico (Vongchan et al., 2009), alimenticio (Shahidi, et al., 1999; Khor, 2001) y farmacéutico (Torrado, 2014; Kato, 2004).
A pesar de la diversidad de métodos reportados para la determinación del grado de N-acetilación del quitosano, el método más referenciado es la correlación desde el espectro infrarrojo de las bandas de vibración, para lo cual la selección de las bandas a correlacionar normalmente involucra una de las bandas amida (I o III) y otra banda que sirva de referencia (Brugnerotto, 2001).
Brugnerotto (2001) y Peniche (2006) han propuesto algunas ecuaciones para el cálculo de la N-acetilación tanto de la quitina como del quitosano, tomando las bandas características de la amida I y como referencia la banda de los grupos hidroxilos, para la correlación lineal propuesta en (1) para la quitina, y para las bandas características de la amida III y como referencia los grupos metilos, para la correlación lineal propuesta en (2) para el quitosano.

Según Ming-Tsung et al. (2009), el grado de N-desacetilación en la quitina estará dado por la ecuación (3).
Grado de DA(%) = 100 - grado de N acetilación (3)
Sin embargo, este método no es del todo eficaz y genera altos costos de inversión en la caracterización de estos biopolímeros a escala de laboratorio.
Con el objetivo de disminuir los costos de caracterización del quitosano y determinar el porcentaje de desacetilación, surgen algunas técnicas, entre las cuales la viscosimetría y la potenciometría son las más simples, rápidas y de bajo costo, requiriendo equipamiento y reactivos fácilmente disponibles en cualquier laboratorio.
Por medio de un ensayo de viscosimetría se puede calcular el peso molecular del quitosano extraído. Para implementar dicha técnica es necesario utilizar diferentes definiciones de viscosidad para transformar la viscosidad cinemática (medida) en la viscosidad reducida, ya que a partir de esta última se puede determinar la viscosidad intrínseca y el peso molecular. Para llevar a cabo este procedimiento se utilizan las definiciones presentadas en las ecuaciones 4 a 8.

Conociendo la viscosidad intrínseca se puede determinar el peso molecular en las muestras analizadas, siempre y cuando el polímero obedezca la ecuación de Huggins; esto es, si presenta un comportamiento lineal entre la concentración y la viscosidad reducida:

En las ecuaciones 6, 7 y 8 el parámetro C hace referencia a la concentración de quitosano en la solución.
La viscosidad intrínseca mide el volumen específico efectivo de un polímero aislado, razón por la que su determinación se realiza extrapolando a concentración nula. Su valor depende del tamaño y forma de la molécula de soluto, asícomo de su interacción con el solvente y de la temperatura de trabajo.
Para un sistema polímero-solvente puede utilizarse la expresión de Mark-Houwink (9) para determinar el peso molecular promedio del polímero (Parada, 2004; Kasaai, 2007).
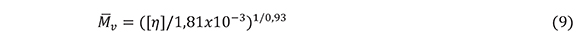
La valoración potenciométrica, por su parte, consiste en disolver el quitosano en un exceso de ácido clorhídrico (HCl) para que ocurra la protonación del grupo amino libre del quitosano y luego realizar una valoración con hidróxido de sodio (NaOH) hasta que el pH de la solución se estabilice, lo cual permite obtener una curva de titulación del quitosano, la cual ha de presentar dos puntos de inflexión, y la diferencia entre estos proporciona la razón de la cantidad de ácido requerido para protonar los grupos amino del quitosano (Hidalgo, 2008); la concentración de estos se puede determinar mediante la expresión (10):

Donde: y es el punto de inflexión mayor y x el menor (expresados como volúmenes)
f es la molaridad de la solución de NaOH
w es la masa en gramos de la muestra
16,1 es un factor asociado al tipo de proteína en estudio
El objetivo del presente trabajo es desarrollar un método que facilite la caracterización del quitosano, al encontrar su peso molecular y determinar la relación entre el porcentaje del grupo amino y el grado de desacetilación del quitosano utilizando las técnicas de viscosimetría y potenciometría.
2. Materiales y métodos
Para el desarrollo de este artículo se emplearon muestras de quitosano de tres fuentes diferentes: una muestra de quitosano extraído del hongo Ganoderma lucidum, una muestra de quitosano extraído de caparazón de crustáceos, ambas producidas en el laboratorio de Biomateriales de la Universidad de Antioquia, y una tercera muestra de quitosano de caparazón de crustáceos grado comercial (SIGMA- ALDRICH >75% desacetilado), utilizado para fines de comparación con las dos muestras obtenidas en el laboratorio.
2.1 Caracterización de las muestras de quitosano
2.1.1 Difracción de Rayos X (XRD)
La cristalinidad de las tres muestras de quitosano evaluadas, al igual que la presencia de fases contaminantes provenientes de las fuentes de extracción, se analizó por medio de difracción de rayos X (XRD), para lo cual se utilizó un difractómetro de rayos X marca RIGAKU tipo miniflex con radiación de ánodo de cobre monocromático, filtro de níquel y detector proporcional (CuKα, λ= 1,5418Å), empleando un rango de 2θ entre 5° y 50°, con un paso de 2º/ min, un voltaje de 30 KV y 20 mA.
2.1.2 Espectroscopía de Infrarrojo con Transformada de Fourier (FTIR)
Los grupos funcionales característicos de las muestras de quitosano evaluadas fueron evaluados en un espectrofotómetro de infrarrojo marca MATTSON 5000, calibrado con una pastilla de bromuro de potasio (KBr), trabajado para un rango de frecuencia entre 450 y 4000 cm-1.
2.1.3 Determinación del porcentaje de N-Acetilación desde los espectros infrarrojos
Para determinar el grado de acetilación de los biopolímeros de quitosano se realizó inicialmente un estudio cuantitativo a partir del área bajo las curvas de los espectros de infrarrojo obtenidos, a cada uno, siguiendo el procedimiento de algunos investigadores como Brugnerotto [12], Peniche [13] y Ming-Tsung [14].
2.2 Determinación del peso molecular mediante la viscosimetría
Para determinar el peso molecular de las muestras de quitosano se utilizó un viscosímetro Brookfield con volúmenes de 500 mL a temperatura ambiente en promedio (25ºC) utilizando una aguja número 1 y una velocidad de giro a 100 RPM. Las muestras de quitosano se prepararon por disolución en una mezcla de ácido acético 0,1 M y cloruro de sodio 0,2 M. Se trabajó con varias concentraciones de quitosano (5,6x10-4 g/mL, 6,6x10-4 g/mL y 8,0x10-4 g/mL).
Una vez se establecieron todas las condiciones de trabajo, se procedió a determinar el tiempo de caída de las disoluciones poliméricas, y con los datos conseguidos se procedió a graficar los puntos correspondientes a las viscosidades reducidas con respecto a la concentración, y se tomó una regresión lineal de estos a partir de las ecuaciones anteriormente planteadas, para determinar la viscosidad intrínseca y posteriormente el peso molecular.
2.3 Determinación del porcentaje de N-Acetilación mediante la potenciometría
Para determinar el contenido de grupos amino de las distintas muestras de quitosano, se utilizó un potenciómetro Hanna Instruments. Inicialmente se diluyeron 0,5 g de quitosano en 20 mL de ácido clorhídrico 0,3 M, posteriormente se tituló la mezcla con una solución de NaOH 0,1 M midiendo el cambio de pH por cada 2 mL de base añadida, manteniendo agitación continua; finalmente se construyó una curva de titulación de pH vs. mL de NaOH añadido para calcular el grado de N-desacetilación del quitosano empleando la ecuación 10. Los mismos procedimientos se realizaron para todas las muestras de quitosano utilizadas.
3. Resultados y discusión
3.1 Caracterización de las muestras de quitosano
3.1.1 Difracción de Rayos X (XRD)
Las Figuras 1, 2 y 3 presentan los difractogramas de las diferentes muestras de quitosano evaluadas, del quitosano extraído de caparazón de crustáceos, del quitosano extraído del hongo Ganoderma lucidum y del quitosano comercial, respectivamente.
Figura 1. Difracción de rayos X para el quitosano extraído de caparazón de crustáceos.
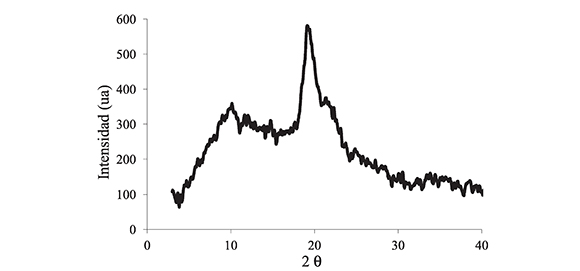
Figura 2. Difracción de rayos X para el quitosano extraído del hongo Ganoderma lucidum.

Figura 3. Difracción de rayos X para el quitosano comercial (SIGMA).

Los difractogramas de las Figuras 1 y 3, correspondientes a fuentes de crustáceos y comercial, respectivamente, presentan los dos picos característicos de un biopolímero de quitosano semicristalino, para un pico principal a 20 = 19,5° y un pico secundario en 20 = 10°, mientras que el espectro de la Figura 2, correspondiente al quitosano extraído de hongo, solo exhibe el pico principal a 20 = 19,5° mostrando menor cristalinidad en la muestra, evidenciado por una menor intensidad y un mayor ancho de banda.
Si bien los espectros de los quitosanos sintetizados difieren en intensidad, comparados con los presentados en los mismos ángulos por el quitosano comercial, en ninguno de los espectros se evidencia la presencia de fases diferentes a las del biopolímero de interés. Específicamente, el espectro del quitosano obtenido desde el caparazón de crustáceos no muestra ninguna fase que pueda ser atribuida a la presencia de calcio, resultado de una deficiente desmineralización, lo que indica que las concentraciones y los tiempos empleados en los procesos parecen ser adecuados.
Sin embargo, todas las muestras de quitosano obtenidas tienen espectros similares a los reportados en la literatura [5, 6, 13], por lo que se puede tener confiabilidad de que es un biopolímero de buenas características para ser evaluado.
3.1.2 Espectroscopía de Infrarrojo con Transformada de Fourier (FTIR)
En las Figuras 4, 5 y 6 se aprecian los espectros infrarrojos obtenidos de las muestras de quitosano sintetizado desde los caparazones de crustáceos, quitosano obtenido del hongo Ganoderma lucidum y quitosano comercial, respectivamente.
Figura 4. Espectro de infrarrojo del quitosano extraído de caparazón de crustáceos.

Figura 5. Espectro de infrarrojo del quitosano extraído del hongo Ganoderma lucidum
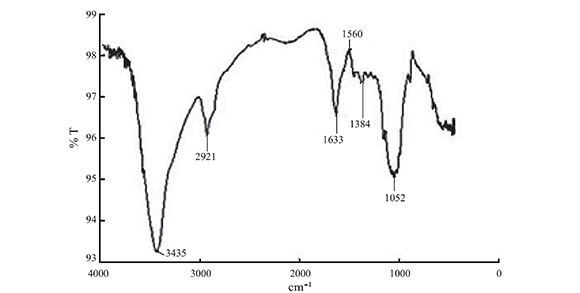
Figura 6. Espectro de infrarrojo del quitosano comercial (SIGMA)

En todos los espectros de las Figuras 4, 5 y 6 se observan las bandas de absorción típicas del quitosano en casi los mismos valores de frecuencia, que concuerdan con las bandas de absorción reportadas por Paulino et al. (2006), Brugnerotto et al. (2001) y Peniche (2006), en sus diferentes reportes de quitosano extraídos de diversas fuentes.
La banda más representativa del biopolímero está ubicada en aproximadamente 3430 cm-1 (3429 cm-1 para el quitosano de crustáceos y 3435 cm-1 para el quitosano de hongos y comercial). Esta banda corresponde al estiramiento N-H para la amida I y banda ancha de vibración tensión de los grupos hidroxilo.
Se puede apreciar también una banda muy cercana a 2920 cm-1, atribuida a la vibración tensión del C-H; y una banda ubicada en 1647 cm-1 para el quitosano de crustáceos corresponde al estiramiento C=O de la amida I y la observada a 1633 cm-1 en la muestra de quitosano de hongos y comercial, puede ser atribuida a la vibración de estiramiento C-N de los grupos superpuestos C=O también de la amida I.
La banda ubicada en 1560 cm-1 es una banda de estiramiento o deformación de la amida II, mientras que la banda ubicada cerca de 1380 cm-1 corresponde a la deformación simétrica de los grupos CH3 y la ubicada en 1052 cm-1 representa el enlace débil C-N y banda C-O.
La banda ubicada en 1318 cm-1 en el espectro de la Figura 4 corresponde a la deformación CO-NH al grupo CH2 de la amida III, pero a juzgar por su apariencia, podría también deberse a humedad en la muestra.
3.1.3 Determinación del porcentaje de N-Acetilación desde los espectros infrarrojos
Para la determinación del porcentaje de N-acetilación conseguido para el quitosano de las diferentes fuentes, con la correlación lineal expresada en (2), se tomó como banda característica la amida III localizada a 1318 cm-1 y como referencia la banda de grupos metilos a 1380 cm-1, y la desacetilación se tomó como la resta del grado de N-acetilación, expresada en (3).
Los porcentajes de N-acetilación y de desacetilación calculados desde (2 y 3) para el quitosano de crustáceos son:

Los valores calculados a partir de las ecuaciones arrojan un porcentaje de desacetilación del 80,67% para el quitosano extraído de caparazón de crustáceos, acorde con lo reportado por Peniche (2006).
Los porcentajes de N-acetilación y desacetilación calculados de (2 y 3) para el quitosano de hongos son:

Como se puede observar en los resultados anteriores, se tiene que la desacetilación del quitosano obtenido a partir de hongos se encuentra apenas por debajo del límite inferior del rango esperado, esto puede deberse a que los valores de las absorbancias tomados no corresponden estrictamente a 1318 cm-1 y 1380 cm-1 como lo pide la correlación empírica sino los valores de 1462 cm-1 y 1380 cm-1 que son los que más se aproximan (Figura 5).
Para el quitosano comercial se tomó como banda característica la amida III localizada a 1376 cm-1 y como referencia la banda de la amida secundaria a 1561 cm-1. A partir de esto, los porcentajes de N-acetilación y desacetilación del quitosano comercial son:

A partir de los resultados obtenidos para los tres quitosanos evaluados, se observa que el grado de desacetilación de las muestras se encuentra dentro del rango esperado (70 - 85%), lo que corrobora que las muestras obtenidas desde los caparazones de crustáceos y desde el hongo Ganoderma lucidum son en efecto el polímero de interés.
3.2 Determinación del peso molecular mediante la viscosimetría
En la Tabla 1 se aprecian los resultados de los ensayos de viscosimetría realizados por triplicado a las diferentes muestras de quitosano evaluadas, luego de aplicar las ecuaciones (4 a 8) presentadas.
Tabla 1. Ensayos de viscosimetría realizados a las muestras de quitosano

En la Figura 7 se observan las regresiones lineales de los puntos de viscosidad reducida en función de la concentración para las muestras de quitosano evaluadas (viscosidad reducida vs concentración).
Figura 7. Regresión lineal de los puntos de viscosidad en función de la concentración en las muestras de quitosano

De las curvas de la Figura 7 se evidencia que la viscosidad reducida de las muestras de quitosano analizadas presenta un comportamiento lineal con respecto a la concentración del polímero en la solución de ácido acético y cloruro de sodio. Además se observa que el coeficiente de regresión está muy próximo a un valor de 1, lo cual indica que es válido afirmar que el polímero sintetizado cumple con la ecuación de Huggins.
Además del comportamiento lineal de la viscosidad de la solución de quitosano, se puede inferir que la viscosidad se encuentra directamente relacionada y la relación es proporcional a la concentración del polímero en la solución, es decir, a menor cantidad de polímero, la solución obtenida será menos viscosa, y a medida que dicha concentración se incrementa la viscosidad se debe incrementar. Estos resultados van acorde a lo esperado según estudios previos realizados a este tipo de biopolímero (Tsaih & Chen, 1999).
A continuación, las ecuaciones para la regresión de los datos de los ensayos realizados a los quitosanos derivados de hongos y crustáceos y al quitosano comercial, con su respectivo coeficiente de regresión:

En la Tabla 2 se presentan las viscosidades intrínsecas para todos los ensayos y los valores de los respectivos pesos moleculares obtenidos.
Tabla 2. Resultados de viscosidad y peso molecular para las muestras de quitosano evaluadas
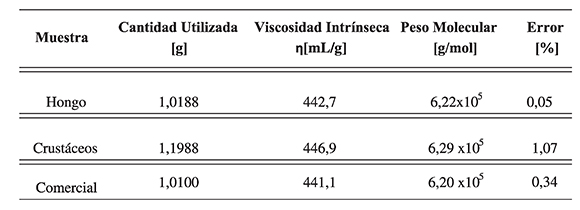
En general, para los ensayos realizados con los quitosanos sintetizados en el laboratorio y el quitosano comercial, los resultados de viscosidad intrínseca fueron muy similares. Asímismo, el peso molecular obtenido también se encuentra dentro de los rangos reportados para el quitosano. La literatura reporta que el peso molecular del quitosano debe estar aproximadamente en 6,22x105 g/mol (Parada et al., 2004); y a partir del cálculo del error porcentual se verifica que los resultados para el cálculo del peso molecular son muy precisos, por lo cual puede afirmarse que los polímeros presentan las características reportadas para el quitosano.
3.3 Determinación del porcentaje de N-Acetilación mediante la potenciometría
En la Figura 8(a) se muestran los resultados de la valoración potenciométrica para la muestra de quitosano extraído de Ganoderma lucidum, donde se genera una curva de titulación con dos puntos de inflexión, cuyos valores se determinaron según el criterio de la primera derivada; dichos puntos corresponden a los máximos graficados en la Figura 8(b).
Figura 8. Curva de titulación el quitosano extraído de Ganoderma lucidum (a) y criterio de la primera derivada (b)

Similarmente, en la Figura 9(a) se muestran los resultados de la valoración potenciométrica para la muestra de quitosano extraído de caparazones de crustáceos, donde se generan curvas de titulación con dos puntos de inflexión, cuyos valores se determinaron según el criterio de la primera derivada; dichos puntos corresponden a los máximos graficados en la Figura 9(b).
Figura 9. Curva de titulación para el quitosano extraído crustáceos (a) y criterio de la primera derivada (b)

Finalmente, en la Figura 10(a) se exhiben los resultados de la valoración potenciométrica para la muestra de quitosano comercial y los máximos correspondientes a sus puntos de inflexión se observan en la Figura 10(b).
Figura 10. Curva de titulación para el quitosano comercial (a) y criterio de la primera derivada (b).

En la Tabla 3 se muestran los valores correspondientes a los máximos graficados en las Figuras 8(b), 9(b) y 10(b), además de los porcentajes de desacetilación calculados empleando (10).
Tabla 3. Proporción de grupos amino en las muestras de quitosano mediante titulación potenciométrica

Si bien el grado de desacetilación reportado por el fabricante se encuentra por encima del 75% para la muestra de quitosano comercial, el porcentaje de grupos amino obtenidos experimentalmente se encuentra dentro de los márgenes aceptables para este ensayo, según el rastreo bibliográfico realizado. Las muestras de quitosano obtenido en el laboratorio, por su parte, exhiben un comportamiento similar al observado en los resultados para el quitosano comercial y las proporciones de grupo amino obtenidos para estas muestras se encuentran dentro de márgenes aceptables.
4. Conclusiones
Con los resultados obtenidos de las caracterizaciones por difracción de Rayos X y espectroscopía de infrarrojo, se comprobó que el material de partida para análisis potenciométrico corresponde a quitosano y no a quitina.
El peso molecular de los quitosanos obtenidos en el laboratorio tiene valores de 6,29x105 g/mol y 6,22x105 g/mol, los cuales se encuentran dentro de los márgenes reportados para este biopolímero, por lo cual se puede afirmar que los protocolos de fabricación de quitosano que se están implementando en el laboratorio son adecuados y, en efecto, se está obteniendo quitosano y no alguno de sus derivados como el quitano.
El grado de desacetilación obtenido experimentalmente tanto para las muestras de quitosano comercial como para las de quitosano sintetizado de diferentes fuentes se encuentra dentro de los márgenes aceptables para este ensayo, comparado con la técnica desarrollada desde los espectros infrarrojos.
La proporción del grupo amino promedio para la serie de datos estudiada es de 62,3%, lo cual permite establecer que la prueba es repetible y puede aplicarse a diferentes muestras de quitosano bajo las condiciones que dispone el Laboratorio de Biomateriales de la Universidad de Antioquia.
Los resultados arrojados para las muestras de quitosano estudiadas permiten clasificarlo como quitosano de aplicabilidad industrial debido a que se encontraron porcentajes de grupos aminos muy similares a los obtenidos para muestras de quitosano comercial, y dentro de los límites establecidos en la literatura para este material.
De los datos tomados se logra obtener una tendencia de la relación entre proporción de grupo amino y grado de desacetilación que tiende a ser directamente proporcional, debido a que para mayor proporción de grupo amino se evidencia la presencia de un mayor grado de desacetilación.
Agradecimientos
Los autores agradecen al Laboratorio de Bioprocesos de Ingeniería Química y al Laboratorio de Biomateriales del Programa de Bioingeniería de la Universidad de Antioquia por la ayuda con los equipos y las muestras facilitadas para el desarrollo de este artículo y a la estudiante Carolina Marín por la ayuda prestada.
Referencias
Brugnerotto, J., Lizardib, J., Goycoolea, F., Argüelles, W., & Desbrières, J. (2001). An infrared investigation in relation with chitin and chitosan Characterization. Polymer, 42(8), 3569-3580. [ Links ]
Chandumpai, A., Singhpibulporn, N., & Sornprasit, P. (2004). Preparation and physicochemical characterization of chitin and chitosan from the pens of the squid species, Loligo lessoniana and Loligo formosana. Carbohydrate Polymers, 58, 467-474. [ Links ]
Fernández, M., Heinämäki, J., Räsänen, M., Maunu, S., Karjalainen, M., Nieto, et. al. (2004). Solid-state characterization of chitosan derived from lobster chitin. Carbohydrate Polymers, 58 (4), 401-408. [ Links ]
Hernández, H., Águila, E., Flores, O., Viveros, E., y Ramos, E. (2009). Obtención y caracterización de quitosano a partir de exoesqueletos de camarón. Superficies y Vacío, 22 (3), 57-60. [ Links ]
Hidalgo, C., Suárez, Y., y Fernández, M. (2008). Validación de una técnica potenciométrica para determinar el grado de desacetilación de la quitosana. Ars Pharmaceutica, 49 (3), 245-257. [ Links ]
Hirano, S. (1999). Chitin and chitosan as novel biotechnological materials. Polymer International, 48 (8), 732-734. [ Links ]
Kasaai, M. (2007). Calculation of Mark-Houwink-Sakurada (MHS) equation viscometric constants for chitosan in any solvent-temperature system using experimental reported viscometric constants data. Elservier Carbohydrate Polimers, 68 (3), 477-488. [ Links ]
Kato, Y., Onishi, H., y Machida, Y. (2004). N-succinyl-chitosan as a drug carrier: water-insoluble and water-soluble conjugates. Biomaterials, 25 (5), 907-915. [ Links ]
Khor, E. (2001). Chitin: fulfilling a biomaterials promise. New York, USA: Elsevier. [ Links ]
Ming-Tsung, Y., Joan-Hwa, Y., & Jeng-Leun,M. (2009). Physicochemical characterization of chitin and chitosan from crab shells. Carbohydrate Polymers, 75 (1), 15-21. [ Links ]
Parada, L., Crespín, G., Miranda, R., y Katime, I. (2004). Caracterización de quitosano por viscosimetría capilar y valoración potenciométrica. Revista Iberoamericana de Polímeros, 5 (1), 1-16. [ Links ]
Paulino, A., Simionato, J., Garcia, J., & Nozaki, J. (2006). Characterization of chitosan and chitin produced from silkworm chrysalides. Carbohydrate Polymers, 64, 98-103. [ Links ]
Peniche, A. (2006). Estudios sobre quitina y quitosana. Tesis doctoral, Universidad de la Habana, La Habana, Cuba. [ Links ]
Shahidi, F., Kamil, J., & Jeon, Y. (1999). Food applications of chitin and chitosans. Trends in Food Science & Technology, 10 (2), 37-51. [ Links ]
Torrado, S., Prada, P., & De la Torre, P. (2004). Chitosan-poly(acrylic) acid polyionic complex: in vivo study to demonstrate prolonged gastric retention. Biomaterials, 25 (5), 917-923. [ Links ]
Tsaih, M., & Chen, R. (1999). Effects of ionic strength and pH on the diffusion coefficients and conformation of chitosans in molecule solution. Journal of Applied Polymer Science, 73 (10), 2041-2050. [ Links ]
Vongchan, P., Sajomsang, W., Subyen, D. y Kongtawelert, P. (2003). Anticuagulant activities of the chitosan polysulfate synthesized from marine crab by semi heterogeneous conditions. Science Asia, 29 (11), 115-120. [ Links ]
Yamaguchi, I., Itoh, S., Suzuki, M., Sakane, M., Osaka, A., & Tanaka, J. (2003). The chitosan prepared from crab tendon I: the characterization and the mechanical properties. Biomaterials, 24 (12), 2031-2036. [ Links ]

Revista de Ciencias por Universidad del Valle se encuentra bajo una licencia Creative Commons Reconocimiento 4.0.

