










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Cirugía
Print version ISSN 2011-7582On-line version ISSN 2619-6107
rev. colomb. cir. vol.22 no.3 Bogotá July/Sept. 2007
Seguimiento de una familia
Follow to family
(1) Residente de cirugía general. Universidad Pontificia Bolivariana. Medellín, Colombia.
(2) Profesor titular de patología, Universidad Pontificia Bolivariana. Patólogo, Hospital Pablo Tobón Uribe y Dinámica IPS. Medellín, Colombia.
(3) Research Fellow. Molecular and Population Genetics Lab. London Research Institute. Cancer Research. Londres - Reino Unido.
(4) Coloproctólogo. Hospital Pablo Tobón Uribe. Medellín, Co- lombia.
(5) Cirujano general y laparoscopista. Hospital Pablo Tobón Uribe. Medellín, Colombia.
(6) Cirujano endoscopista. Hospital Pablo Tobón Uribe. Medellín, Colombia.
(7) Internista gastroenterólogo. Hospital Pablo Tobón Uribe. Medellín, Colombia.
(8) Cirujano gastroenterólogo. Hospital Pablo Tobón Uribe. Medellín, Colombia.
Correspondencia: María Helena Gaitán Buitrago, MD. Correo electrónico: mhg78cx@gmail.com Medellín, Colombia
Fecha de recibo: Noviembre 10 de 2006. Fecha de aprobación:Mayo 2 de 2007
Resumen
El síndrome Peutz - Jeghers es una enfermedad con pólipos del colon con un patrón autosómico dominante, su tipo histológico es hamartomatoso y aunque es muy rara la transformación maligna, se ha postulado una secuencia hamartoma - carcinoma. Este artículo describe el seguimiento de un caso índice y su familia.
Palabras clave: síndrome de Peutz-Jeghers, cáncer colorrectal, pólipos del cólon, poliposis familiar del colon.
Abstract
Peutz - Jeghers syndrome is a polyposis syndrome that appears to be inherited as an autosomal dominant gene; the polyps are hamartomatous, and although malignant transformation is rare, a hamartoma-carcinoma sequence has been postulated. This article describes an index case and his family follow up.
Key words: Peutz-Jeghers syndrome, colorectal, neoplasms, colonic polyps, familial polyposis coli.
Introducción
El término pólipo se refiere a una protuberancia anormal de una superficie epitelial. Los pólipos son usualmente asintomáticos pero se pueden ulcerar y sangrar, debutar con dolor abdominal y cuando son muy grandes producir obstrucción intestinal. Hay numerosos tipos de pólipos colorrectales, que pueden clasificarse como mucosos y submucosos (1).
Los pólipos mucosos pueden ser neoplásicos y no neoplásicos. Dentro de los pólipos neoplásicos benignos se incluyen los adenomas y según su patrón histológico son tubulares, vellosos o tubulovellosos. Los pólipos con patrón histológico de malignidad presentan carcinoma in situ, carcinoma invasivo o carcinoma polipoide. Los no neoplásicos incluyen los pólipos hiperplásicos, juveniles, Peutz-Jeghers, pólipos inflamatorios, o los que son repliegues de epitelio normal (tabla 1). Los pólipos submucosos se originan en varios tipos de tejidos e incluyen lipomas, leiomiomas, colitis cística profunda, entre otras (tabla 2). Cripps en 1881 fue el primero en describir los aspectos heredofamiliares de los pólipos gastrointestinales.


Reporte del caso
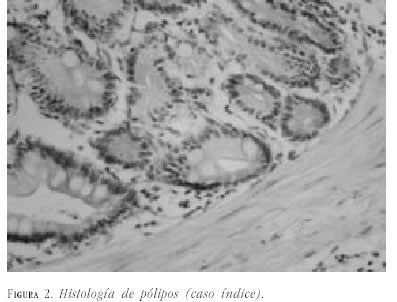
Se trata de un paciente de sexo masculino de años 63 edad, procedente de San Pedro de los Milagros (Antioquia), que consulta por dos meses de evolución de dolor epigástrico urente, pérdida de peso y vómito. Al examen físico se encontró hiperpigmentación en piel de plantas de pies, palmas de manos, labios y mucosa oral. Se hospitalizó para identificar la causa del dolor abdominal y dentro de los estudios se encontró intususcepción intestinal por lo que se llevó a cirugía; en el acto operatorio se halló una masa de aspecto tumoral con múltiples pólipos sésiles y pediculados en la luz del colon, entre 0.5 y 3.5 cm y pedículos de 1 a 6 cm de longitud (figura 1); se resecó un segmento de 68 cm de hemicolon izquierdo, cuyo reporte histológico fue adenocarcinoma mucosecretor y pólipos hamartomatosos (figura 2). El paciente no dio información fehaciente para determinar si hubo casos similares en sus padres o hermanos o si él fue el primer caso de la enfermedad.

Se realizó seguimiento de su familia, compuesta por el paciente, su esposa y 10 hijos, de los cuales 4 eran hombres y 6 mujeres; dos de sus hijos habían fallecido (figura 3). Uno de los hijos de la familia falleció a los 31 años de edad por obstrucción intestinal pero no fue clara la causa y no se estudió para síndromes polipósicos.
El hijo mayor, a los 36 años de edad consultó al hospital por astenia, adinamia, vértigo y melenas. Presentaba pigmentación café oscura en los labios. Durante una gastroscopia para identificar el origen de las melenas se observaron múltiples pólipos en fondo y cuerpo gástricos, de tamaño variable y cuya histología reportó pólipo hamartomatoso. En una colonoscopia se detectaron más pólipos con histología hamartomatosa; con estos datos se hizo el diagnóstico de síndrome de Peutz-Jeghers (SPJ).
La segunda hija fue hospitalizada por pérdida de peso, melenas e intususcepción intestinal. Se realizó extracción de pólipos por cecostomía y sigmoidostomía con resección de 10 cm de íleon. La patología reportó pólipo mixto de 2.5 cm con patrón hiperplásico y adenomatoso, sin malignidad y múltiples pólipos hiperplásicos en íleon sin malignidad.
La tercera hija de la familia consultó por dolor abdominal. Con la presencia de manchas hiperpigmentadas en labios y el antecedente familiar de Peutz-Jeghers se realizó endoscopia digestiva superior y resección intestinal que evidenció múltiples pólipos hiperplásicos gástricos y yeyunales. Durante el seguimiento se encontró cáncer de colon que fue manejado quirúrgicamente pero falleció. Dos de sus hijas fueron evaluadas por presentar manchas hiperpigmentadas en los labios y episodios intermitentes de sangrado digestivo. A ambas se les han realizado resecciones intestinales y reintervenciones por bridas.
El menor de los hijos del caso índice presenta desde la infancia las lesiones hiperpigmentadas en labios (figura 4). A los 26 años se le resecaron múltiples pólipos hamartomatosos en estómago, íleon, ciego y colon descendente. Desde entonces se han realizado colonoscopias periódicamente para evaluar la progresión de la enfermedad y practicar polipectomías (figura 5). Hasta el momento no se han encontrado lesiones malignas.


Discusión
El síndrome Peutz-Jeghers pertenece a los síndromes de pólipos hamartomatosos heredados. La asociación entre pólipos gastrointestinales y las lesiones mucocutáneas fue descrita por Peutz en 1921, posteriormente Jeghers en 1949 describió el patrón de transmisión genética. Estos pólipos son lesiones hamartomatosas de epitelio glandular soportadas por células de músculo liso contiguas a la muscular de la mucosa. Los pólipos son usualmente benignos pero pueden crecer progresivamente y producir síntomas o desarrollar transformación maligna. Su transmisión es autosómica dominante por mutaciones en el gen STK11 en el cromosoma 19p13.3 cerca al marcador D19S886. El gen STK11/LKB1 codifica una serina/treonina kinasa que se expresa en todos los tejidos humanos. Este gen sufre una baja frecuencia de mutaciones en el síndrome de Peutz-Jeghers (SPJ), y se ha propuesto que los sitios de mutación ocurren en los exones 1 a 6 los cuales provocan en conjunto la mitad de las alteraciones germinales descritas. De cualquier manera, no todas las familias con SPJ están ligadas al locus 19p13.3 del gen STK11/LKB1, sugiriendo heterogenicidad. El posible potencial maligno de los pólipos hamartomatosos en el SPJ no está bien entendido. Aunque es muy rara, la transformación maligna de los pólipos del SPJ ha sido reportada lo que indica la posibilidad de una secuencia hamartoma-carcinoma; sin embargo, las mutaciones en el gen K-ras son menos frecuentes que en la FAP. Las diferencias en las alteraciones moleculares genéticas entre la secuencia adenomacarcinoma y los tumores relacionados con el SPJ sugieren una diferente vía de carcinogénesis (7).
El SPJ se presenta en uno de cada 120.000 nacimientos. Hasta 6% de los hamartomas removidos de los pacientes pueden presentar cambios adenomatosos.
Se caracteriza por manchas pigmentadas en los labios, mucosa oral, plantas de pies y palmas de manos, acompañados de múltiples pólipos hamartomatosos gastrointestinales; 65 a 95% en el intestino delgado, 60% en el colon y 50% en el estómago. La aparición de los síntomas suele ser en la adultez temprana; presentan intususcepción del intestino delgado, obstrucción y sangrado. El riesgo relativo para desarrollar cáncer de cualquier sitio es hasta 15 veces mayor que en la población general, principalmente en colon y recto, páncreas, mama, ovarios y endometrio.
El manejo puede incluir la resección endoscópica de los pólipos por enteroscopia intraoperatoria o colonoscopia. La colectomía se reserva para los casos asociados a cáncer de colon pero no como profilaxis (1-17).
Referencias
1. Lawrence SP, Ahnen DJ. Approach to the patient with colonic polyps. Uptodate 11.2. [ Links ]
2. Seitz U, Bohnacker S, Soehendra N. Endoxcopic Removal. Uptodate 11.2. [ Links ]
3. Bonis PAL, Ahnen DJ. Screening. Uptodate 11.2. [ Links ]
4. Itzkowitz SH, Kim YS. Colonic polyps and polyposis syndromes. En: Sleisenger & Fordtran's Gastrointestinal and Liver Disease. 6 edition. [ Links ]
5. Rustgi AK. Hereditary gastrointestinal polyposis and nonpolyposis syndromes. N Engl J Med 1994; 331 (25): 1694-1702. [ Links ]
6. Read TE, Read JD, Butterly LF. Importance of adenomas 5 mm or less in diameter that are detected by sigmoidoscopy. N Engl J Med 1997; 336 (1): 8-12. [ Links ]
7. Entius M, Keller JJ, Westerman AM, et al. Molecular genetic alterations in hamartomatous polyps and carcinomas of patients with Peutz-Jeghers syndrome. J Clin Pathol 2001; 54: 126-131. [ Links ]
8. Chung DC, Mino M, Shannon KM. A 45-Year-Old woman with a family history of colonic polyps and cancer. N Engl J Med 2003; 349 (18): 1750-1760. [ Links ]
9. Bond JH. Polyp Guideline: Diagnosis, treatment, and surveillance for patients with colorectal polyps. Practice Guideline November 2000; 95 (11): 3053-3063. [ Links ]
10. Keller JJ, Offerhaus GJA, Drillenburg P, et al. Molecular analysis of sulindac-resistant adenomas in familial Adenomatous Polyposis. Clin Cancer Res 2001; 7: 4000-4007. [ Links ]
11. McGarrity TJ, Kulin HE, Zaino RJ. Peutz-Jeghers syndrome. Am J Gastroenterol 2000; 95 (3): 596-604. [ Links ]
12. Restrepo C, Moreno J, Duque E, et al. Juvenile colonic polyposis in Colombia. Dis Col & Rect 1978; 21 (8): 600-612. [ Links ]
13. Entius MM, Westerman AM, Giardiello FM. Peutz-Jeghers polyps, dysplasia, and K-ras codon 12 mutations. GUT 1997; 41: 320-322. [ Links ]
14. Doxey BW, Kuwada SK, Burt RW. Inherited polyposis syndromes: molecular mechanisms, clinicopathology, and genetic testing. Clin Gastroenterol Hepatol Jul 2005; 3 (7): 633-641. [ Links ]
15. Soares J, Lopes L, Vilas Boas G, Pinho C. Wireless capsule endoscopy for evaluation of phenotypic expression of small-bowel polyps in patients with Peutz-Jeghers syndrome and in symptomatic first-degree relatives. Endoscopy Dec 2004; 36 (12): 1060-1066. [ Links ]
16. Pennazio M. Small-bowel endoscopy. Endoscopy. Jan 2004; 36 (1): 32-41. [ Links ]
17. Yee NS, Furth EE, Pack M. Clinicopathologic and molecular features of pancreatic adenocarcinoma associated with Peutz-Jeghers syndrome. Cancer Biol Ther. Jan-Feb 2003; 2 (1): 38-47. [ Links ]

