











 Portuguese (pdf)
Portuguese (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUÇÃO
As doenças raras de maneira geral acometem um pequeno número de indivíduos, porém, coletivamente, representam um problema significativo de saúde pública, seja por seus aspectos clínicos específicos seja pelas tecnologias em saúde que demandam. Grande parte dessas doenças ainda carecem de tratamentos aprovados e por isso, vários países como dos Estados Unidos da América (EUA) e países-membros da União Europeia (UE), já estabeleceram caminhos normativos e incentivos regulatórios e econômicos específicos visando mudar esse contexto e catalisar o desenvolvimento dessas tecnologias monitorado por seus órgãos sanitários (Food and Drug Administration (FDA) e European Medicines Agency (EMA), respectivamente) [1, 2].
A principal estratégia global de fomento é a regulação de registro dos chamados "medicamentos órfãos" destinados ao diagnóstico, prevenção ou tratamento dessas doenças e que contam com diferentes experiências internacionais com destaque para EUA e UE [1, 3].
Os marcos regulatórios dos EUA (Orphan Drug Act-1983) (4) e da UE (Orphan Drug Regulation-2000) [5] normatizam ao apoio à pesquisa e desenvolvimento de medicamentos órfãos, por meio de diferentes incentivos, inclusive econômicos, como: exclusividade de marketing (10 anos na UE e sete anos nos EUA), redução nos custos de desenvolvimento clínico (nos EUA há redução de até 50% de créditos fiscais), subvenções para pesquisa clínica, redução de taxas regulatórias (redução da taxa de 40% na UE-gratuita em algumas situações) [1, 6].
Incentivos econômicos e flexibilização dos critérios e exigências regulatórias para esse tipo de medicamento é o que mais tem sido defendido pelas indústrias farmacêuticas e associações ligadas a doenças raras. Cabe referir que, nos últimos anos, especialmente no contexto internacional, as indústrias farmacêuticas têm se empenhado cada vez mais no desenvolvimento de medicamentos órfãos como resultado dos estímulos já referidos e avanços tecnológicos relacionados, além do grande potencial de geração de receita resultante de preços elevados associados a alguns desses medicamentos [7, 8].
O avanço normativo no âmbito internacional remeteu à discussão sobre a temática no Brasil onde a iniciativa regulatória relacionada se deu efetivamente em 2017 pela Agência Nacional de Vigilância Sanitária (Anvisa) com a proposição da Resolução da Diretoria Colegiada-RDC n° 205 [9]. Contudo a norma brasileira não regulamentou como as demais agencias o registro específico de medicamentos órfãos e apenas deixou a possibilidade de dispensar estudos de fase Ill para doenças raras, "quando a realização destes estudos não for viável" [9].
Registrar esses medicamentos no país é sobretudo uma forma de monitorar seu uso. Isso é importante pois há incertezas de efetividade terapêutica e segurança decorrentes de estudos frágeis e grande potencial de impacto econômico dos medicamentos para doenças raras no sistema de saúde. O paradoxo regulatório desse tipo de medicamento está justamente no equilíbrio entre o incentivo ao desenvolvimento de tecnologias com ampliação do seu acesso [7, 10, 11].
Deve-se considerar a potencialidade que a escassez de mecanismos regulatórios e políticas públicas específicas tem em termos negativos quanto ao acesso, custos associados e ao monitoramento da efetividade e segurança quando do uso desses medicamentos [12]. Ademais, a comparação relativa entre os medicamentos utilizados por pacientes com doenças raras e as principais doenças e agravos em nível local e global é fundamental [13] na perspectiva de prospecção de valor atribuído ao uso desses medicamentos desde sua pesquisa e desenvolvimento do ponto de vista do paciente e do sistema de saúde em termos de sustentabilidade [14].
Assim, este artigo buscou investigar os medicamentos com registro ativo no Brasil que se enquadram internacionalmente como medicamentos órfãos com vistas a explorar possíveis impactos gerados pela ausência de uma normativa sanitária específica sobre essas tecnologias no país.
METODOLOGIA
Este estudo envolveu uma abordagem exploratória dividida em duas etapas.
A primeira etapa baseou-se identificação dos medicamentos registrados como órfãos na UE [15, 16] e dos EUA [17, 18]. Foram coletadas nas bases das respectivas agencias informações sobre (i) nome genérico/substância ativa, (ii) nome comercial, (iii) apresentações disponíveis e (iv) indicações clínicas aprovadas. Foram considerados os itens com registro ativo até dezembro de 2019.
Foram incluídos na análise os medicamentos de origem farmoquímica, natural (vegetal, animais e mineral), biotecnológica (genômica, genômica funciona, proteômica, meta-bolômica e citômica) e de química sintética com informações (i-iv) completas. Foram desconsiderados os medicamentos com dados incompletos ou indisponíveis. Ao final dessa etapa obteve-se uma lista de referência internacional dos medicamentos órfãos.
Na segunda etapa, essa lista foi comparada com base de registros de medicamentos da Anvisa [19] para identificação dos itens com registro ativo no Brasil. Para isso foram também considerados apenas os medicamentos com registro ativo em 2019 e utilizado como referencia a Denominação Comum Brasileira (DCB) [20] com intuito de evitar equívocos de descrição dos itens.
Os dados foram consolidados utilizando o programa Excel®. Os medicamentos foram agrupados de acordo com as categorias terapêuticas, de acordo com o Anatomical Therapeutic Chemical Classification System (ATC) [21]. As indicações de uso dos medicamentos foram classificadas de acordo com a Classificação Internacional de Doenças (CID-10) [22] e relacionadas a dados de carga de doença (global e nacional) conforme os três grandes grupos (i) doenças transmissíveis, maternas, neonatais e nutricionais, (ii) doenças crônicas não transmissíveis, (iii) causas externas, considerando o componente-chave da estimativa de carga de doença analisado foi de anos vividos com incapacidade, do inglês Disability Adjusted Life Years (DALY) [23].
RESULTADOS
Foram identificados 369 medicamentos órfãos registrados nos órgãos europeu e estadunidense, dos quais 351 estavam registrados no FDA e 208 na EMA (190 com registro ativo nos dois órgãos). Essa diferença pode se dar pelo maior tempo de regulamentação nos EUA e pelo registro de alguns itens no EMA em fluxo tradicional.
Metade dos medicamentos órfãos registrados (49,6 %) se enquadram em 2 grupos ATC era de agentes antineoplásicos e imunomoduladores (n=135; 36,59 %) e de medicamentos que agiam no aparelho digestivo e metabolismo (n=48; 13,01 %), tabela 1.
Tabela 1 Fármacos registrados como medicamentos órfãos nos órgãos FDA e EMA por grupo

ATC: Anatomical Therapeutic Chemical Classification System; EMA: European Medicines Agency; FDA: Food and Drug Administration.
Dos elencos de medicamentos órfãos registrados como tal no âmbito internacional, 177 não apresentavam registros ativos no Brasil (47,97 %). As classes que mais obtiveram itens sem registro também foram os antineoplásicos e imunomoduladores (n=45), seguido de Aparelho digestivo e metabolismo (n=29). As maiores proporções intragrupo sem registro no Brasil foram dos medicamentos que agem em órgãos dos sentidos (90 %), no aparelho geniturinário, hormônios sexuais (83 %) e medicamentos dermatológicos (80 %), como pode ser observado no gráfico 1.
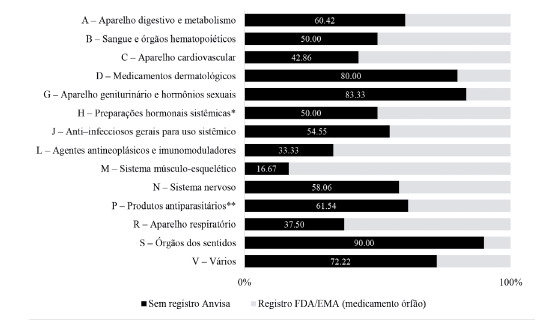
*Excluindo hormônios sexuais e insulinas; **Mais inseticidas e repelentes. Anvisa: Agência Nacional de Vigilância Sanitária; EMA: European Medicines Agency; FDA: Food and Drug Administration.
Gráfico 1 Proporção (%) de medicamentos com registros como medicamentos órfãos na FDA e EMA, mas sem registro no Brasil por grupo ATC (2019).
Os medicamentos órfãos registrados na FDA e na EMA tinham 801 indicações clínicas potenciais no total, sendo a maioria delas para o tratamento de neoplasias (tumores) e doenças endócrinas, nutricionais e metabólicas que juntam representaram pouco mais da metade das indicações analisadas (N=426; 52 %), tabela 2.
Tabela 2 Indicações dos medicamentos órfãos registrados na FDA/EMA.

CID: Classificação Internacional de Doenças; EMA: European Medicines Agency; FDA: Food and Drug Administration (FDA).
Das indicações clínicas dos medicamentos órfãos com registro nos órgãos internacionais, 327 (40,82 %) envolviam medicamentos sem registro ativo no Brasil. As indicações com maior escassez de registro foram as neoplasias (93) seguido das doenças endócrinas, nutricionais e metabólicas (68). Na comparação intragrupo ocorreu maior percentual de ausência de registro nas doenças dos sentidos e doenças do aparelho geni-turinário, como pode ser observado no gráfico 2.

Das 327 indicações clínicas de medicamentos não registrados no Brasil, a maioria (N=283; 86,54 %) era pertencente ao grupo de causas de óbito de doenças não transmissíveis cuja carga de doenças é maior, o que pode sinalizar que mesmo que inicialmente os itens sejam registrados para tratar doenças mais raras, podem possuir tendência de aplicação futura em doenças e públicos maiores, tabela 3.
Tabela 3 Indicações de medicamentos órfãos registrados na FDA/EMA e sem registro no Brasil e carga local e global de doença por grupos de causas de óbito.
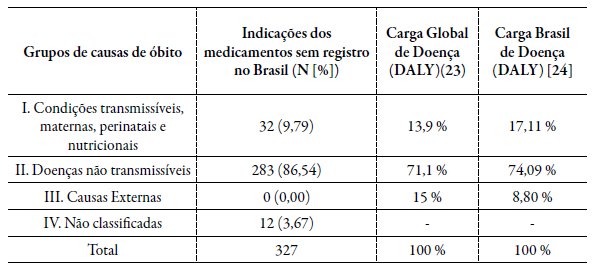
DALY: Estimativa de anos de vida ajustados por incapacidade (do inglês, Disability adjusted life years); EMA: European Medicines Agency; FDA: Food and Drug Administration.
DISCUSSÃO
Apesar da haver variação quanto à definição de doenças raras entre os países, elas correspondem, do ponto de vista epidemiológico, a doenças com baixa incidência e prevalência. No entanto, que em uma perspectiva coletiva, as doenças raras têm características heterogêneas e, juntas, correspondem a uma carga global de doença significativa [1, 25]. Grande parte dessas doenças tem origem essencialmente genética acometendo pacientes pediátricos contando com um bom número de estudos que visam identificar potenciais causas e melhorar seu rastreamento e diagnóstico [26]. Entretanto, esse avanço relativo do conhecimento relacionado à temática não tem se traduzido em pesquisa e desenvolvimento de tecnologias em saúde com finalidade terapêutica em uma taxa proporcional [1, 12].
Dessa forma, a abordagem sobre medicamentos órfãos torna-se fundamental em um cenário de discussão sobre seu acesso somando o contexto da falta de regulamentações claras acerca do tema em vários países, como o Brasil [25]. A comparação com as políticas relacionadas a esses medicamentos em países onde já há instituição de incentivos para a indústria como os EUA e países-membro da UE é importante do ponto de vista prático visando à melhoria do seu acesso considerando a necessidade de racionalizar o uso de recursos e tomadas de decisões, além da promoção de avanços normativos e de acesso ao mercado, com regras e incentivos claros [25].
Nos EUA e na UE, regulamentos específicos foram implementados há anos visando fornecer incentivos para desenvolvimento e ampliação do acesso de medicamentos órfãos [27, 28]. Somado a isso, o estabelecimento de redes globais de doenças raras que apoiam o desenvolvimento de medicamentos, os avanços tecnológicos na área e a mudança de paradigma na pesquisa e desenvolvimento de medicamentos órfãos em termos de receita gerada pelas indústrias farmacêuticas resultaram em crescimento da oferta de medicamentos órfãos no âmbito internacional [3, 7, 29-31]. Isso, por sua vez, tem impactado em termos de registros desses medicamentos nos órgãos reguladores
A falta de regulação clara e específica que incentive à pesquisa, desenvolvimento e registro desses medicamentos tende a impactar diretamente o acesso aos pacientes brasileiros. Evidenciado com quase metade dos itens observados nesse estudo sem registro ativo no país com três centenas de indicações potencialmente importantes para doenças com poucas alternativas disponíveis. Necessário frisar que alguns itens identificados possuem registro com apresentações e indicações limitadas, a exemplo da Hidroxiureia que possui registro no Brasil apenas do comprimido de 500 mg, de uso adulto e para uso em neoplasias, já nos EUA há disponível concentrações menores (100 mg, 200 mg, 400 mg) e para uso também na Doença Falciforme, doença negligenciada e tratada de forma off-label e adaptada no Brasil [32].
A doença falciforme é congênita e como outras tem se destacado como alvo de registro de medicamentos órfãos internacionalmente. Outro exemplo verificado é o caso do Ibuprofeno injetável, usado na neonatologia para Ducto arterial patente, um problema cardiológico importante que tem sido frequente com o aumento dos nascimentos prematuros. Seu uso precoce pode estar associado a redução de casos cirúrgicos. Mais uma vez observa-se o registro do fármaco no Brasil, mas a exclusão de uma forma farmacêutica especifica voltada a casos mais especializados. O próprio Ministério da Saúde Brasileiro já apontou que o publico pediátrico é o mais prejudicado com a ausência de normas regulatórias que facilitem o registro de medicamentos para doenças raras e negligenciadas [33, 34].
Como observado nesse estudo, há uma grande tendência internacional em relação ao registro de algumas doenças, como as oncológicas e as metabólicas [1, 27, 30]. Isso se deve essencialmente a interesses comerciais que vem influenciando consideravelmente o desenvolvimento de medicamentos para tratamento de determinados grupos de doenças raras cujo foco envolve alternativas terapêuticas que deem maior rentabilidade [29, 30].
Essas doenças representam uma grande carga para o paciente e para a família/ cuidadores, cujo impacto geralmente não é levado em consideração nas análises de custo-efetividade padrão e mesmo com grande interesse mercadológico, quando os mecanismos de registro são complexos é comum que haja desestimulo ao registro em países de renda média ou em desenvolvimento [12, 14].
Apesar do grande número de indicações voltados as doenças raras, é necessário frisar que na análise de carga global de doença (DALY) focada em doenças crônicas não transmissíveis como as neoplasias pode demonstrar tendência de que esses medicamentos possam no futuro ter ampliação de uso de canceres mais raros, para canceres mais frequentes por exemplo [24]. Entretanto, observa-se também que alguns que teriam uma menor carga de doença nos EUA ou na Europa, representam um grande problema de saúde publica no Brasil, como por exemplo a Miltefosina, registrado como a medicamento órfão no FDA e atualmente importado sem registro na Anvisa para uso no SUS por se tratar do único medicamento oral disponível para leishmaniose [35-37].
Trata-se de um medicamento adquirido mesmo sem registro, porém uma exceção a regra, pois no Brasil uma licença válida é requisito fundamental para a comercialização ao publico ou aquisição e distribuição pelo sistema de saúde. O grande número de itens sem registro identificados nesse estudo aponta para uma maior pressão por importações particulares excepcionais e fomento a judicialização de medicamentos. O que além de gerar inequidades de acesso, dificulta o processo inclusive de monitoramento da segurança de uso comum num processo após registro [28, 36, 38].
Um esforço de inovação normativa no Brasil e uma harmonização com regras sobre medicamentos órfãos internacionais podem favorecer inclusive a colaboração entre centros de pesquisa, indústrias farmacêuticas, representantes de pacientes, autoridades sanitárias e provedores de assistência à saúde para o enfrentamento às doenças raras ou doenças negligenciadas no país [29-31, 39, 40].
CONCLUSÃO
O equilíbrio entre a pesquisa e desenvolvimento de medicamentos e a geração de evidências científicas robustas deve ser mediado pela construção de aspectos regulamentares específicos que potencialmente viabilizarão o acesso a medicamentos ditos seguros do ponto de vista clínico. Isso remete, portanto, à necessidade clara de evolução normativa quanto aos medicamentos para tratamento de doenças raras no Brasil em contraposição ao já estabelecido internacionalmente em países desenvolvidos.
O envolvimento dos diversos atores de maneira integrada é fundamental no que se refere à criação e implementação efetiva de programas e políticas específicas que remetam a recursos de informação, pesquisas básicas, atendimento clínico, incentivos à pesquisa e ao desenvolvimento de medicamentos que garantam os direitos de acesso à assistência à saúde dos pacientes com doenças raras no Brasil.

