










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Reumatología
Print version ISSN 0121-8123
Rev.Colomb.Reumatol. vol.14 no.4 Bogotá Oct./Dec. 2007
Artículo de Revisión
Manifestaciones osteoarticulares por amiloidosis sistémica
Osteoarticular manifestations by systemic amyloidosis
Carlos-Enrique Toro Gutiérrez1, Mario-Andrés Quintana Duque2, José Félix Restrepo3, Federico Rondón4, Oscar Páez5, Antonio Iglesias-Gamarra6
Recibido para publicación: septiembre 24/2007 Aceptado en forma revisada: noviembre 20/2007
Resumen
La amiloidosis es un grupo de enfermedades cuyo común denominador es el depósito extracelular de fibrillas insolubles derivadas de proteínas en órganos y tejidos. De acuerdo a su etiología y al tipo de proteína depositada existen varias clases de amiloi-dosis. A pesar que la incidencia de amiloidosis sisté -mica secundaria (AA) ha disminuido notoriamente con el advenimiento de drogas modificadoras de la enfermedad (DMARD) y terapia biológica, continúa siendo el tipo de amiloidosis más frecuentemente observada por el reumatólogo. En este artículo revisamos la historia, clasificación, epidemiología, diagnóstico y tratamiento de la amiloidosis sistémica haciendo énfasis en las manifestaciones osteoar-ticulares que produce la enfermedad y en las distintas enfermedades reumatológicas que pueden originar una amiloidosis secundaria (AA). Así mismo publicamos un material fotográfico recopilado durante 20 años en diferentes centros de reumatología del país que es de gran ayuda para realizar el diagnóstico clínico de esta infrecuente patología.
Palabras clave: amiloidosis sistémica, artritis reumatoide, inflamación
Summary
Amyloidosis is a generic term that refers to the extracellular tissue deposition of fibrils composed of low molecular weight subunits of a variety of proteins. Amyloidosis classification depends on its etiology and subtype of protein involved. Systemic secondary amyloidosis (AA) is the most frequent subtype seen on rheumatology services because rheumatoid arthritis is currently the most frequent cause of AA, although its incidence has been declined because a better treatment of rheumatoid arthritis with disease-modifying anti-rheumatic drugs (DMARD). In this review we provide a general overview of the pathogenesis, clinical manifestations, diagnosis, and treatment of the systemic amyloidosis, emphasizing on the rheu-matic manifestations of these disorders. Besides, we present a photographic material obtained in the last 20 years in several rheumatologic centers in our country that it has a crucial role in the diagnosis and follow-up of this infrequent pathology.
Key words: systemic amyloidosis, rheumatoid arthritis, inflammation.
Introducción
Amiloidosis se refiere a un término genérico que designa el depósito extracelular de fibrillas compuestas de subunidades de bajo peso molecular (5 a 25 kD) de una variedad de proteínas. Al menos 25 diferentes precursores de proteína amiloide se han identificado en humanos. El tipo de fibrilla depende del tipo de amiloidosis, siendo las más importantes los fragmentos de cadenas livianas kappa o lambda de inmunoglobulinas (Ig) en la amiloidosis sistémica primaria (AL) o; los fragmentos de proteína amiloide sérica A en la amiloidosis sistémica secundaria (AA), la cual está asociada con trastornos inflamatorios crónicos o; la ß2-microglobulina en la amiloidosis asociada a diálisis.
Los depósitos amiloides pueden presentarse en cualquier órgano, incluyendo el corazón, riñones, tracto gastrointestinal, sistema músculoesquelético y sistema nervioso central (SNC).
Historia
Rudolph Virchow en 1854 adoptó el término amiloide, inicialmente introducido por Schleiden en 1838 con el objetivo de describir el almidón, para referirse a la presencia de depósitos tisulares de una sustancia semejante a la celulosa cuando se expone al yodo. No obstante, existen algunas descripciones de casos del siglo XVII que bien podrían corresponder a amiloidosis. En 1639 Nicolaus Fontanus realizó lo que probablemente corresponda a la primera descripción de amiloidosis al realizar la autopsia de un hombre que murió de ascitis e ictericia. El hígado de este individuo tenía un absceso y una gran hepatomegalia. Posteriormente, en 1657, Bartholin describe el bazo de una mujer en su libro titulado Historiarum Anatomicarum Rariorum Centuria cuyas características macroscópicas eran como carne dura. En 1679 Theophile Bonet, un médico de Ginebra, en su obra Sepulchretum Sive Anatomia Practica recopiló, sin mayor discriminación, todas las observaciones anatomo-patológicas que encontró a la fecha. Entre las 3000 autopsias descritas incluyó los dos casos anteriores, creyéndose actualmente que fueron los primeros casos descritos de amiloidosis. En estas descripciones se observó que los depósitos amiloides tenían un aspecto ceroso macroscópicamente, pero hialino y amorfo cuando se observaba por microscopía de luz. Probablemente Wilks, en 1865, fue quien informó inicialmente la amiloidosis reactiva en un paciente al parecer con osteomielitis crónica, aun cuando se le acredita a Wild, en 1886, la descripción de la amiloidosis primaria. Weber en 1903 informó la asociación con el mieloma múltiple. Posiblemente, el primer caso de enfermedad articular amiloidea fue expuesto por Buch, en 1873. Benhold en 1922 introduce la prueba de Rojo Congo, para detectar la presencia de amiloidosis, aun en ausencia de signos clínicos. Magnus Levy en 1931, fue el primero en apreciar la frecuencia con la cual las proteínas de Bence Jones se asociaban a la amiloidosis; previamente, Lubarsch, en 1929, describe tres casos de amiloidosis primaria y plantea los criterios clínicos para diferenciarla de la secundaria. La primera mención de compromiso óseo en la amiloidosis difusa fue descrita por Gerber en 1934. En 1972, Wiernik hace una recopilación de 40 casos de compromiso articular por amiloide1 . El análisis por microscopía electrónica de depósitos amiloides, se realizó por primera vez en 1959; mostraba usualmente fibrillas de 8 a 10 mm de ancho, rectas y no ramificadas, las cuales eran compuestas por microfilamentos cuando se analizaban a una mayor resolución2,3.
A nivel nacional y latinoamericano, los informes sobre este tópico son escasos, pero vale la pena resaltar los estudios de Maldonado y colaboradores4, en 1965, quienes diferencian el plasmocito de maduro e inmaduro y esto, realmente, pone en evidencia la diferencia entre las gammapatías benignas y malignas de acuerdo con las características de anaplasia del plasmocito. Ordóñez5, Piñeros y DArchiardi6 publicaron un caso de amiloidoma como tumor primario de costilla. Posteriormente Iglesias1, en 1986, reportó seis casos de amiloidosis sistémica, entre los que se incluía una paciente con síndrome POEMS asociado. No se han publicado más series de casos describiendo las manifestaciones de la amiloidosis sistémica en Colombia. Tan solo Marin et al.7 informaron las características de dos pacientes con cardiopatía amiloidea en 2001.
Clasificación
Amiloidosis sistémica primaria (AL): es una dis-crasia de células plasmáticas que da lugar a una proliferación clonal de dichas células en la médula ósea. Las células plasmáticas producen depósitos fibrilares cuyo principal componente son subtipos de cadenas livianas kappa y lambda derivados de inmuno-globulinas. La mayoría de los estudios relacionan la subclase lambda a los casos de AL, en contraposición a los pacientes con mieloma múltiple que presentan con mayor frecuencia las cadenas kappa. Aunque sus manifestaciones iniciales más frecuentes son fatiga y pérdida de peso, la miocardiopatía y el compromiso neuropático son los rasgos predominantes de este tipo de amiloidosis8.
Amiloidosis sistémica secundaria (AA): es un trastorno caracterizado por el depósito extracelular de fibrillas compuestas por fragmentos de proteína amiloide sérica A (SAA), un reactante de fase aguda. La amiloidosis (AA) puede ser secundaria a un gran número de enfermedades inflamatorias crónicas incluyendo la artritis reumatoide, artritis idiopática juvenil, espondilitis anquilosante, fiebre mediterránea familiar, infecciones crónicas y neoplasias AA. El órgano que con mayor frecuencia se afecta en amiloidosis (AA) es el riñón (~80%). Usualmente se presenta un depósito glomerular amiloide, ocasionándole al paciente el síndrome nefrótico9. No obstante, puede haber otras formas de compromiso renal10,11.
Amiloidosis relacionada a diálisis: es un tipo de amiloidosis asociada al depósito de ß2-microglobulina. Su prevalencia alcanza el 80% en pacientes con diálisis por más de 15 años. Se caracteriza por un compromiso osteoarticular importante y está más frecuentemente asociado con hemodiálisis que con diálisis peritoneal. Aunque se cree que su etiología está relacionada con el estímulo del sistema inmune por el contacto repetido de la sangre con membranas artificiales que genere elevados niveles de ß2-microglobulina, el hecho de que se presente en pacientes con diálisis peritoneal e incluso en pacientes con insuficiencia renal crónica que no han requerido terapia dialítica, sugiere que el compromiso renal crónico per se juega un papel relevante en su patogénesis12.
Amiloidosis familiar: constituye un grupo de enfermedades autosómicas dominantes que se caracterizan por el depósito extracelular de fibrillas de amiloide en varios órganos y tejidos, siendo predominante la afectación de los nervios periféricos. La más común es causada por mutaciones de transti-rretina, una proteína intermediaria en el transporte de las hormonas tiroideas y el retinol. Se han identificado más de 70 mutaciones en diferentes aminoácidos como posibles causantes de la enfermedad, siendo la más común la sustitución de valina por metionina en la posición 3013.
Epidemiología
La incidencia de amiloidosis AA en estudios de autopsia varía de 0.50 a 0.86%14,15. Esta incidencia ha disminuido de manera considerable con el advenimiento en reumatología de drogas modificadoras de la enfermedad y terapia biológica. Antes de 1990, cuando tan solo se contaba con AINES y corticoides para controlar las enfermedades autoinmunes, no era infrecuente observar casos de amiloidosis (AA), principalmente asociados a artritis reumatoide. De acuerdo con estadísticas norteamericanas la AR y la AIJ continúan explicando el mayor número de casos de amiloidosis (AA) asociado a enfermedad reumato-lógica con un 48-56% del total. Espondilitis anquilosante (5-8%), artritis psoriática (4-5%) y fiebre familiar del Mediterráneo (2-3%), aunque en menor medida, también se asocian al desarrollo de amiloidosis (AA)16.
En otras partes del mundo esta tendencia puede variar, encontrando síndromes febriles periódicos como el TRAPS (TRAPS del inglés, tumor necrosis factor receptor-1-associated periodic syndrome) e infecciones en mayor proporción17.
Etiología
Artritis reumatoide (AR): la incidencia post-mortem de amiloidosis complicando la AR en el adulto varía desde un 10 a 25%18-22, aunque estudios previos sugieren un compromiso cercano al 60%23.
La amiloidosis (AA) ocurre con mayor frecuencia en pacientes con enfermedad mal controlada, severa, seropositiva, extraarticular y de larga evolución. De acuerdo a diferentes registros se considera que el intervalo promedio entre el inicio de AR y el desarrollo de amiloidosis (AA) es cercano a 15 años24. El compromiso más frecuente en estos pacientes se encuentra a nivel renal.
La presencia de amiloidosis en AR es indicativa de muy mal pronóstico. Se considera que el diagnóstico de amiloidosis AA acorta la expectativa de vida por cerca de siete años en pacientes con AR.
Artritis idiopática juvenil (AIJ): la prevalencia de amiloidosis (AA) varía del 1-10% en pacientes con JIA, siendo el subtipo de inicio sistémico el más frecuentemente relacionado con el desarrollo de la enfermedad. Similar a AR, el compromiso renal es el más importante y frecuente25.
Espondilitis anquilosante (EA): la EA es una causa menos frecuente de amiloidosis (AA). Igualmente, el compromiso renal es el más importante de la enfermedad26,27.
Lupus eritematoso sistémico (LES): la amiloi-dosis (AA) ha sido descrita en una pequeña proporción de pacientes con LES28. La razón de una menor incidencia no se conoce completamente pero parece relacionada con una menor elevación de SAA como reactante de fase aguda29. Otra explicación propuesta indica que existe una mayor predilección para que SAA circulante presente proteólisis en AR que en LES30.
Otras enfermedades reumáticas: existen informes que relacionan la amiloidosis (AA) con otras enfermedades reumáticas, incluyendo síndrome de Reiter31, enfermedad de Behcet32, arteritis de Takayasu33, enfermedad de Whipple34, polimialgia reumática/arteritis de células gigantes35-37, gota38, enfermedad de Still del adulto39-40 RS3PE41 y síndrome de Sjögren42.
Manifestaciones musculoesqueléticas de la amiloidosis sistémica
Dentro de las manifestaciones musculoesqueléticas de la amiloidosis puede existir compromiso de músculos, articulaciones, huesos y estructuras periarticulares.
Compromiso muscular: la infiltración amiloide de los músculos puede causar aumento de su tamaño (pseudohipertrofia), lo cual se puede observar hasta en el 25% de los casos43. Uno de los sitios donde con mayor frecuencia se afecta es la lengua, manifestado por macroglosia y/o indentación lateral de la lengua (Figura 1). Así mismo, se puede observar engrosamiento simétrico de los labios con mucinosis papilar (Figura 2). Usualmente la infiltración muscular es sistémica. En una revisión retrospectiva se identificaron 12 pacientes con miopatía amiloide comprobada por biopsia durante un periodo de 35 años. Una gran proporción de estos pacientes presentaron adicionalmente alteraciones electrofisio-lógicas, reflejando un compromiso neuropático asociado y en general un mal pronóstico44.


Otra importante forma de compromiso muscular es la miopatía amiloide por cuerpos de inclusión, la cual es la causa más común de miopatía metabólica en personas mayores de 50 años. Igualmente puede encontrarse compromiso muscular secundario a neuropatía o angiopatía amiloide caracterizado por debilidad muscular progresiva, atrofia y discapacidad.
Compromiso articular: microdepósitos de fragmentos amiloides han sido encontrados en cápsulas fibrosas, cartílago y membranas sinoviales en diferentes articulaciones45-47. La incidencia de este compromiso osteoarticular varía con la edad del individuo48.
El compromiso articular en amiloidosis (AL) puede involucrar el sinovio e incluso semejar una AR49. No obstante, con mayor frecuencia el compromiso en la artropatía amiloide es subagudo y lentamente progresivo, con una predilección por los hombros, rodillas, muñecas, metacarpofalángicas (MCF) e interfalángicas proximales (IFP), seguido por los codos y caderas. Por lo general existe poca rigidez matutina y las articulaciones son levemente dolorosas, lo cual permite su diferenciación con la AR50, 51. No obstante, en ocasiones el compromiso articular es tan similar a la AR que dificulta su diferenciación de esta entidad, especialmente en algunas ocasiones en que predomina el compromiso articular en manos asociado a rigidez matinal importante, fatiga y nódulos similares a los subcutáneos observados en la AR52. La radiografía de manos ayuda a diferenciar estas dos entidades (Tabla 1) (Figura 3).


El aspirado del líquido sinovial en amiloidosis revela un infiltrado no inflamatorio, con algunas células mononucleares. Adicionalmente puede demostrarse fragmentos amiloides mediante tinción de rojo Congo por análisis del sedimento centrifugado53.
Adicionalmente puede presentarse compromiso periarticular en amiloidosis sistémica por edema de tejidos blandos secundario a hipertrofia nodular del sinovio directamente infiltrada por amiloide. Esto es especialmente importante alrededor de la articulación glenohumeral, resultando en el peculiar signo shoulder pad54, característico de la amiloidosis AL (Figura 4a y 4b ).


Cuando la mano se encuentra comprometida puede observarse nodularidad y engrosamiento de la fascia palmar con contracturas en flexión y marcada debilidad (Figura 5). Este compromiso se asocia frecuentemente con síndrome de túnel del carpo, el cual es típicamente bilateral y simétrico55. La pérdida de sensación en una articulación puede resultar en una artropatía crónica y destructiva. El prototipo de este trastorno fue descrito por Charcot en relación con tabes dorsal.

Osteopatía amiloide: lesiones tumorales solitarias o múltiples pueden ser observadas en pacientes con amiloidosis AL. Estas lesiones usualmente están ocupadas por material amiloide y pueden asociarse a dolor o fracturas patológicas13,14.
Otras manifestaciones: en pacientes con amiloi-dosis AL puede observarse claudicación de mandíbula,
debida a compromiso vascular amiloide de las ramas faciales de la arteria carótida externa56. La evidencia de gamapatía monoclonal y una pobre respuesta a corticoides permite diferenciar la amiloidosis AL de la polimialgia reumática/arteritis temporal57.
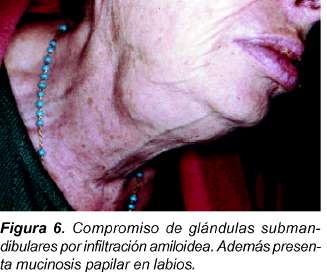
Igualmente se puede presentar un síndrome seco manifestado por xerostomía y xeroftalmia, secundario al depósito amiloide que compromete la función de glándulas salivares y lacrimales58,59 (Figura 6). La biopsia de glándula salivar menor puede ayudar para diferenciar esta entidad del síndrome de Sjögren60. Otra estructura que puede verse infiltrada por el depósito amiloide son los párpados, dando una apariencia característica (Figura 7).

La manifestación más frecuente de la amiloidosis (AA) asociada a enfermedad reumatológica es el compromiso renal observado en artritis reumatoide. Usual-mente se presenta con una proteinuria marcada en rango nefrótico. El diagnóstico se confirma por biop-sia renal en donde se encuentra el depósito amiloide. La estructura renal más comúnmente comprometida es el glomérulo; pero vasos sanguíneos, intersticio y túbulos también pueden verse afectados61. Este compromiso tiene implicaciones importantes en el pronóstico de la enfermedad. Aquellos pacientes con presencia de depósito amiloide exclusivo a nivel glomerular presentan un deterioro rápidamente progresivo de la función renal con necesidad de diálisis en la mayoría de los casos, a diferencia de los pacientes con compromiso netamente vascular donde la función renal no se deteriora significativamente62.
A continuación hacemos mención particular de dos tipos de amiloidosis (AA) por presentar algunas manifestaciones osteoarticulares específicas.
Amiloidosis asociada a mieloma múltiple y POEMS
Las facetas clínicas óseas y articulares de las mielomatosis son tan variables, que muchas veces el clínico o el radiólogo pueden pasarlas desapercibidas y el avance o deterioro que acarrea el depósito de amiloide en estos órganos puede producir contracturas y limitaciones funcionales importantes. La prevalencia de amiloidosis en mieloma múltiple es del 10% y el 2% de los pacientes con mieloma múltiple smoldering (asintomático) van a desarrollar amiloidosis63.
En individuos con mieloma múltiple las estructuras óseas se afectan más que las articulaciones, pero en algunos sujetos las manifestaciones articulares se caracterizan por el depósito de amiloide en las articulaciones y en tejidos blandos para-articulares, ocasionando contractura articular especialmente en los dedos de las manos; así mismo, es frecuente la artritis de distribución similar a la ocasionada por la AR y el signo shoulder pad previamente descrito.
No existen grandes diferencias entre las manifestaciones músculoesqueléticas ocasionadas por mieloma múltiple de las distintas clases de inmuno-globulinas G, A, D y subclases de inmunoglobulinas G1 y G3. Aparte del compromiso articular suele manifestarse por osteopenia con o sin lesiones osteolíticas (Figura 8), quistes subcondrales, lesiones líticas aisladas, fracturas patológicas, inflamación y depósitos nodulares en tejidos blandos64.

Una variante del mieloma múltiple es el síndrome POEMS, término acuñado por Bardwick en 1980 para designar la asociación de polineuropatía, organo-megalia, endocrinopatía, discrasia de células plasmáticas y compromiso cutáneo (Figura 9). Es usual observar lesiones óseas, en forma de esclerosis u osteolisis siendo lo más frecuente un patrón mixto65.

Amiloidosis relacionada a diálisis
Las manifestaciones musculoesqueléticas de la amiloidosis relacionada con diálisis incluyen síndrome de túnel del carpo, periartritis de hombros, artropatía efusiva y lesiones óseas especialmente en columna cervical.
El síndrome de túnel del carpo y el dolor en hombros son las características clínicas más importantes de este tipo de amiloidosis. La prevalencia de este trastorno aumenta proporcionalmente con el tiempo que el paciente se encuentre en diálisis, aunque por histología se han encontrado algunos depósitos amiloides de ß2-microglobulina tan sólo 23 meses después del inicio de la hemodiálisis66 y 21 meses del inicio de diálisis peritoneal67.
Con frecuencia se presenta un compromiso poliarticular, caracterizado por dolor e inflamación de forma simétrica en hombros, muñecas y manos. Usualmente el dolor aumenta con la actividad física, tiende a empeorar en las noches, especialmente luego de la hemodiálisis. El compromiso articular en hombros se asocia con limitación funcional por afección de estructuras ligamentarias adyacentes.
En manos el depósito amiloide puede depositarse en las palmas semejando una masa de tejido subcutánea, que se muestra mejor cuando el paciente extiende la mano. Este signo se conoce como el signo de la cuerda de guitarra, característico de esta entidad. También puede encontrarse artropatía y cambios destructivos en caderas y rodillas por infiltración amiloide en el sinovio, cápsula articular, cartílago y hueso subcondral. Otra manifestación que se puede encontrar son amiloidomas en región glútea y fosa poplítea68.
El compromiso de la columna vertebral es otra característica importante de este tipo de amiloidosis. Las características de este tipo de espondiloartropatía destructiva69 incluyen disminución de espacios intervertebrales con erosiones marginales de los cuerpos vertebrales sin formación de osteofitos. El segmento cervical es el más afectado de toda la columna vertebral, especialmente en los espacios intervertebrales 4-5 y 5-670,71. Adicionalmente se pueden identificar quistes radiolucentes en los cuerpos de la primera y segunda vértebra cervical o en el proceso odontoideo que pueden afectar la integridad de la articulación atlanto-axoidea y cervico-occipital72.
Amiloidosis asociada a otros trastornos reumatológicos
La amiloidosis AA puede presentarse como una complicación de diferentes trastornos hereditarios caracterizados por fiebre periódica. En este grupo se incluye la fiebre familiar del Mediterráneo (FMF)73,74, el TRAPS75, el síndrome Muckle-Wells76, el síndrome de urticaria familiar al frío (FCAS), el síndrome de hi-pergamaglobulinemia D (HIDS)77 y síndromes relacionados con mutaciones del gen que codifica la criopirina77. La incidencia de amiloidosis AA varía considerablemente con cada uno de estos trastornos, encontrándose desde el 2 al 25%.
El síndrome Muckle-Wells y FMF puede ser asociado con episodios recurrentes de sinovitis transitoria, usualmente afectando una sola articulación. Estos episodios típicamente persisten por pocos días. Adicionalmente se puede observar piartrosis en hombros, caderas y ocasionalmente sacroilíacas en pacientes con FMF79-80.
En pacientes con TRAPS se presentan mialgias y artralgias, las cuales pueden ser severas e incapacitantes, involucrando incluso cabeza y cuello81. La artritis no es una manifestación frecuente de este síndrome.
Diagnóstico de amiloidosis
Aunque el diagnóstico de amiloidosis sistémica puede ser sugerido por algunas características clínicas (p.ej. síndrome nefrótico) en pacientes con factores predisponentes, es necesario confirmar el mismo por biopsia tisular que puede ser tomada de la grasa abdominal subcutánea, tejido rectal, médula ósea o de un órgano clínicamente comprometido.
La aspiración de grasa abdominal subcutánea con tinción por rojo Congo y examinación por luz polarizada tiene una sensibilidad de 57 a 85% y una especificidad de 92 a 100% para amiloidosis AA o AL82-85. La sensibilidad de la biopsia rectal se ha informado en un 84%86. En este mismo informe se encontró una sensibilidad del 90% en biopsias de riñón, hígado o de túnel del carpo.
El depósito amiloide parece como un material hialino amorfo en microscopía de luz. Bajo luz polarizada, las fibrillas amiloides se unen al rojo Congo observándose una birrefringencia verde bajo y a tioflavina T produciendo una fluorescencia verde-amarilla intensa (Figura 10). En microscopía electrónica se aprecian fibrillas no ramificadas de 8-10nm de ancho87. En casos especiales, la inmu-nohistología puede ser útil para identificar la subunidad proteica88.

La microscopía de inmunofluorescencia utilizando una proteína anti-AA es positiva en amiloidosis AA. Una tinción negativa para cadenas kappa o lambda ayuda a diferenciar la amiloidosis AL de AA.
Evaluación de laboratorio: salvo por la biopsia, la evaluación de laboratorio en amiloidosis AA posee poco valor. No obstante, la electroforesis de proteínas séricas o la dosificación de inmunoglobulinas son necesarias para excluir amiloidosis AL.
- Gamagrafía con amiloide sérico P: el amiloide sérico P (SAP) es una proteína de significado fisiológico incierto que se une de forma significativa a las fibrillas amiloides, por lo cual se pueden identificar los depósitos amiloides tisulares mediante gamagrafía seguida por inyección de SAP marcado con tecnecio. La sensibilidad de este método es cercana al 90% en amiloidosis AA y AL y cercana al 50% en amiloidosis hereditaria y menor en amiloidosis cardiaca89. La especificidad en todos los casos se aproxima al 93%90. Una desventaja del estudio es el riesgo de infección que acarrea ya que el sustrato es obtenido por medio de donantes de sangre.
En resumen se recomienda realizar la biopsia tisular para confirmar la presencia de amiloide en todos los pacientes con sospecha de amiloidosis AA. Usualmente el estudio inicia con aspiración de grasa abdominal subcutánea por ser segura y menos invasiva. En caso de un resultado negativo y si el índice de sospecha es alto, se recomienda obtener una muestra rectal o de un órgano clínicamente comprometido. Una vez que el depósito amiloide se confirme se debe realizar el análisis por inmuno-fluorescencia o inmunohistoquímica. No obstante, se debe tener en cuenta que la realización de biopsias tisulares a órganos sólidos como el hígado puede ser peligrosa, dado que la amiloidosis en sí se asocia con un alto riesgo de sangrado91. Esta diátesis he-morrágica se presenta como resultado de dos mecanismos: el primero relacionado con la deficiencia del factor X, el cual se adhiere a las fibrillas amiloides en el hígado y el bazo92; el segundo, por disminución en la síntesis de factores de coagulación como resultado de enfermedad hepática avanzada.
Tratamiento
Amiloidosis sistémica primaria (AL): desde el estudio realizado por Kyle y colaboradores92 en 1985 se ha propuesto como el tratamiento estándar de la amiloidosis sistémica la asociación melfalan-corticoesteroides. Dicha asociación demostró disminución en la progresión de la enfermedad y aumento en la sobrevida comparado con colchicina no sólo en ese trabajo sino en otros estudios liderados por el mismo Kyle94 y Skinner95 posteriormente. El melfa-lan es un agente alquilante cuyo mecanismo de acción es formar enlaces covalentes con los ácidos nucleicos, lo cual produce un bloqueo en la replica-ción del DNA y/o ruptura de sus filamentos96.
Básicamente lo que mostraron los estudios es un aumento en la sobrevida aproximadamente un año, lo cual a pesar de ser benéfico, no se constituye en una opción significativa para estos pacientes. Así mismo, se demostró que esta combinación rara vez logra una remisión completa de la enfermedad y tan sólo el 28% obtiene la remisión parcial9. Por tal motivo, en la última década se han explorado nuevas terapias que han mejorado el pronóstico de la enfermedad. Con altas dosis de dexametasona, 40 mg/ día, 12 días al mes por tres ciclos en fase de inducción, la esperanza de vida es de 2,6 años y el 53% de los pacientes obtiene alguna remisión97. Estos resultados mejoran si a dexametasona, incluso con un esquema de inducción menos agresivo, 40 mg/día 4 días al mes, se le adiciona melfalán 0,22 mg/kg. La esperanza de vida se eleva a 5,1 años con una frecuencia de respuesta cercana al 70%98. No hay un aumento en la incidencia de efectos adversos con este último esquema, siendo las infecciones oportunistas y las citopenias las principales reacciones adversas a tener en cuenta.
El esquema que ha demostrado mayor efectividad es el trasplante autólogo de células madre, asociado tanto a melfalán como a altas dosis de dexametasona. Sería ideal ofrecer esta opción terapéutica a todos los pacientes; sin embargo, muchos pacientes con enfermedad avanzada son incapaces de tolerar la terapia, especialmente aquellos con compromiso cardiaco al momento del diagnóstico. La enfermedad renal y/o hepática avanzada también contraindicaría la terapia99.
Amiloidosis sistémica secundaria (AA): en cuanto a la amiloidosis sistémica secundaria (AA), al ser causada por el depósito de fragmentos proteolíticos de un reactante de fase aguda, amiloide sérico A, el cual se eleva por inflamación crónica, lo primordial es disminuir y tratar la enfermedad de base. No existen, por tanto, terapias específicas que hayan demostrado un beneficio consistente. No obstante, recientemente se ha evaluado eprodisato, un compuesto que interfiere con la unión entre proteínas amiloidogénicas y glucosaminoglicanos, evitando en modelos experimentales el depósito de amiloide en los tejidos100, como terapia en AA con compromiso renal. En este estudio se demostró un efecto favorable al retrasar el deterioro en la función renal, aun-
que no tuvo mayor impacto en cuanto a disminución de la proteinuria101. En nuestra experiencia a través de 20 años, la colchicina ha sido el medicamento más utilizado en amiloidosis sistémica secundaria (AA). Aunque en la literatura su uso no está sustentado bajo estudios con buen grado de evidencia, hemos observado en nuestra casuística que después de 12 meses de tratamiento continuo la mejoría es notable. Dados los resultados obtenidos y el bloqueo del depósito de amiloide extracelular y perivascular que produce el medicamento, recomendamos su uso a dosis de 1,5 mg/día en amiloidosis sistémica secundaria (AA).
Conclusión
La amiloidosis sistémica presenta un amplio espectro de manifestaciones clínicas; independiente del tipo de amiloidosis que la produzca, el compromiso osteoarticular se observa en un porcentaje alto de pacientes. Cuando la amiloidosis es secundaria a patología reumatológica refleja mal control de la enfermedad primaria, severidad y larga evolución. Afortunadamente su incidencia ha disminuido, gracias a las drogas modificadoras de la enfermedad (DMARD) y la terapia biológica. La artritis reumatoide es la enfermedad que explica la mayor cantidad de casos de amiloidosis (AA); usualmente se manifiesta por compromiso renal dado por proteinuria en rango nefrótico. Para el diagnóstico de amiloidosis es fundamental la biop-sia tisular que puede ser tomada de la grasa abdominal subcutánea, tejido rectal, médula ósea o de un órgano clínicamente comprometido. Finalmente, la tendencia actual en el tratamiento de amiloidosis sistémica es el uso de inmunosupresión; sin embargo, debido a la imposibilidad de realizar terapias agresivas en nuestro medio como el trasplante autólogo de células madre, recomendamos el uso de colchicina con buenos resultados para el control de la enfermedad.
Referencias
1. Iglesias A. Amiloidosis sistémica adquirida. Estudio de 6 casos y revisión bibliográfica. Biomédica 1986; 1-2: 26-47. [ Links ]
2. Goldsbury C, Green J. Time-lapse atomic force microscopy in the characterization of amyloid-like fibril assembly and oligomeric intermediates. Methods Mol Biol 2005; 299: 103. [ Links ]
3. Cohen AS, Calkins E. Electron microscopic observations on a fibrous component in amyloid of diverse origins. Nature 1959; 183: 1202. [ Links ]
4. Maldonado JE, Bayrd ED, Brown AL. The flaming cell in multiple myeloma. A light and electron microscopy study. Am J Clin Pathol 1965; 44(6): 605-612. [ Links ]
5. Ordóñez N. Amiloidosis. Universitas Médicas 1973; 15: 111-119. [ Links ]
6. Piñeros J, Dáchiardi R. Amiloidoma como tumor primario de costilla. Acta Med Col 1978; 3: 27-31. [ Links ]
7. Marín J, Duque M, Medina L, Uribe W, Velásquez J. Cardiomiopatía amiloidea Rev. colomb. cardiol 2005; 11: 389-396. [ Links ]
8. Falk RH, Comenzo RL, Skinner M. The systemic amyloi-doses. N Engl J Med 1997; (25) 337: 898-909. [ Links ]
9. Yang GC, Gallo GR. Protein A-gold immunoelectron microscopic study of amyloid fibrils, granular deposits, and fibrillar luminal aggregates in renal amyloidosis. Am J Pathol 1990; 137: 1223. [ Links ]
10. Westermark GT, Sletten K, Westermark P. Massive vascular AA-amyloidosis: a histologically and biochemically distinctive subtype of reactive systemic amyloidosis. Scand J Immunol 1989; 30: 60. [ Links ]
11. Hiki Y, Horii A, Kokubo T, et al. A case of rheumatoid arthritis with renal tubular amyloidosis. Nephron 1994; 68: 394. [ Links ]
12. Kleinman KS, Coburn JW: Amyloid syndromes associated with hemodialysis. Kidney Int 1989; 35: 657-675. [ Links ]
13. Santos M, Dias L, Esperanca P. Importancia de la TTR met 30 en el diagnóstico de la polineuropatíaamiloidótica familiar sin antecedentes familiares. Rev Neurol 2000; 30: 929-931. [ Links ]
14. Simms RW, Prout MN, Cohen AS. The epidemiology of AL and AA amyloidosis. Baillieres Clin Rheumatol 1994; 8: 627. [ Links ]
15. Hazenberg BP, van Rijswijk MH. Where has secondary amyloid gone? Ann Rheum Dis 2000; 59: 577. [ Links ]
16. Gertz MA, Kyle RA. Secondary systemic amyloidosis: Response and survival in 64 patients. Medicine 1991; 70: 246. [ Links ]
17. McAdam KP, Raynes JG, Alpers MP, et al. Amyloidosis: a global problem common in Papua New Guinea. P N G Med J 1996; 39: 284. [ Links ]
18. Gomez-Casanovas E, Sanmarti R, Sole M, et al. The clinical significance of amyloid fat deposits in rheumatoid arthritis: a systematic long-term followup study using abdominal fat aspiration. Arthritis Rheum 2001; 44: 66. [ Links ]
19. Okuda Y, Takasugi K, Oyama T, et al. Amyloidosis in rheumatoid arthritis: Clinical study of 124 histologically proven cases. Ryumachi 1994; 34: 939. [ Links ]
20. Wakhlu A, Krisnani N, Hissaria P, et al. Prevalence of secondary amyloidosis in Asian North Indian patients with rheumatoid arthritis. J Rheumatol 2003; 30: 948. [ Links ]
21. El Mansoury TM, Haszenberg BP, El Badawy SA, et al. Screening for amyloid in subcutaneous tissue of Egyptian patients with rheumatoid arthritis: clinical and laboratory characteristics. Ann Rheum Dis 2002; 61: 42. [ Links ]
22. Wiland P, Wojtala R, Goodacre J, Szechinski J. The prevalence of subclinical amyloidosis in Polish patients with rheumatoid arthritis. Clin Rheumatol 2004; 23: 193. [ Links ]
23. Husby G. Amyloidosis and rheumatoid arthritis. Clin Exp Rheumatol 1985; 3: 173. [ Links ]
24. Myllykangas-Luosujarvi R, Aho K, Kautiainen H, Hakala M. Amyloidosis in a nationwide series of 1666 subjects with rheumatoid arthritis who died during 1989 in Finland. Rheumatology (Oxford) 1999; 38: 499. [ Links ]
25. Woo P. Systemic juvenile idiopathic arthritis: diagnosis, management, and outcome. Nat Clin Pract Rheumatol 2006; 2(1): 28-34. [ Links ]
26. Gratacos J, Orellana C, Sanmarti R, et al. Secondary amyloidosis in ankylosing spondylitis. A systematic survey of 137 patients using abdominal fat aspiration. J Rheumatol 1997; 24: 912. [ Links ]
27. Kovacsovics-Bankowski M, Zufferey P, So AK, Gerster JC. Secondary amyloidosis: a severe complication of ankylosing spondylitis. Two case-reports. Joint Bone Spine 2000; 67: 129. [ Links ]
28. Queffeulou G, Berenbaum F, Michel C, et al. AA amyloidosis in systemic lupus erythematosus: an unusual complication. Nephrol Dial Transplant 1998; 13: 1846. [ Links ]
29. Pepys MB. Serum C-reactive protein, serum amyloid P-component and serum amyloid A protein in autoimmune disease. Clin Immunol Allergy 1981; 1: 77. [ Links ]
30. Migita K, Eguchi K, Tsukada T, et al. Increased circulating Serum Amyloid A protein derivatives in Rheumatoid Arthritis patients with Secondary Amyloidosis. Lab Invest 1996; 75: 371. [ Links ]
31. Anderson CJ, Gregory MC, Groggel GC, Clegg DO. Amyloidosis and Reiters syndrome: report of a case and review of the literature. Am J Kidney Dis 1989; 14: 319. [ Links ]
32. Melikoglu M, Altiparmak MR, Fresko I, et al. A reappraisal of amyloidosis in Behcets syndrome. Rheumatology (Oxford) 2001; 40: 212. [ Links ]
33. Wada Y, Nishida H, Kohno K, et al. AA amyloidosis in Takayasus arteritis-long-term survival on maintenance haemodialysis. Nephrol Dial Transplant 14: 2478. [ Links ]
34. Cruz I, Oliveira AP, Godinho-Lopes JM, et al. Whipples disease and renal amyloidosis. Am J Gastroenterol 1993; 88: 1954. [ Links ]
35. Escriba A, Morales E, Albizua E, et al. Secondary (AA-type) amyloidosis in patients with polymyalgia rheumatica. Am J Kidney Dis 2000; 35: 137. [ Links ]
36. Moraga, Sicilia JJ, Blanco J, Ubeda I. Giant cell arteritis and renal amyloidosis: report of a case. Clin Nephrol 2001; 56: 402. [ Links ]
37. Medina YF, Martínez JB, Restrepo JF, Federico Rondón F, Iglesias-Gamarra A. Polimialgia reumática como presentación de mieloma múltiple y amiloidosis. Rev Colomb Reumatol 2005;12:269-275. [ Links ]
38. Guma M, Bayes B, Bonet J, Olive A. Gout and secondary amyloid. Clin Rheumatol 1999; 18: 54. [ Links ]
39. Rivera F, Gil CM, Gil MT, et al. Vascular renal AA amyloidosis in adult Stills disease. Nephrol Dial Transplant 1997; 12: 1714. [ Links ]
40. Wendling D, Humbert PG, Billerey C, et al. Adult onset Stills disease and related renal amyloidosis. Ann Rheum Dis 1991; 50: 257. [ Links ]
41. Magy N, Michel F, Auge B, Toussirot E, Wendling D. Amyloid arthropathy revealed by RS3PE syndrome. Joint Bone Spine 2000; 67: 475-477. [ Links ]
42. Wong BC, Wong KL, Ip MSM, et al. Sjögrens syndrome with Amyloid A presenting as multiple pulmonary nodules. J Rheumatol 1994; 21: 165. [ Links ]
43. Gertz MA, Kyle RA. Myopathy in primary systemic amyloidosis. J Neurol Neurosurg Psychiatry 1996; 60: 655. [ Links ]
44. Rubin DI, Hermann RC. Electrophysiologic findings in amyloid myopathy. Muscle Nerve 1999; 22: 355. [ Links ]
45. Ladefoged C, Merrild U, Jorgensen B. Amyloid deposits in surgically removed articular and periarticular tissue. Histopathology 1989; 15: 289. [ Links ]
46. Rumpelt HJ, Braun A, Spier R, et al. Localized amyloid in the menisci of the knee joint. Pathol Res Pract 1996; 192: 547. [ Links ]
47. Cole AS, Cordiner-Lawrie S, Carr AJ, Athanasou NA. Localised deposition of amyloid in tears of the rotator cuff. J Bone Joint Surg Br 2001; 83: 561. [ Links ]
48. Goffin YA, Thoua Y, Potvliege PR. Microdeposition of amyloid in the joints. Ann Rheum Dis 1981; 40: 27. [ Links ]
49. Donnelly S, Bourne JT, Levison DA, et al. Amyloid arthritis associated with IgM kappa lymphoplasmacytoid lymphoma. Br J Rheumatol 1993; 32: 1004. [ Links ]
50. de Ruiter EA, Ronday HK, Markusse HM. Amyloidosis mimicking rheumatoid arthritis. Clin Rheumatol 1998; 17: 409. [ Links ]
51. Fujishima M, Komatsuda A, Imai H, et al. Amyloid arthropathy resembling seronegative rheumatoid arthritis in a patient with IgD-kappa multiple myeloma. Intern Med 2003; 42: 121. [ Links ]
52. Gordon DA, Pruzanski W, Ogryzlow MA, et al. Amyloid arthritis simulating rheumatoid disease in five patients with multiple myeloma. Am J Med 1973; 55: 142. [ Links ]
53. Gordon DA, Pruzanski W, Ogryzlow MA. Synovial fluid examination for the diagnosis of amyloidosis. Ann Rheum Dis 1973; 32: 428. [ Links ]
54. Edelson JG. Amyloid shoulder pads. Two cases of multiple myeloma. Acta Orthop Scand 1995; 66: 292. [ Links ]
55. Kyle RA, Gertz MA. Primary systemic amyloidosis: Clinical and laboratory features of 474 cases. Semin Hematol 1995; 32: 45. [ Links ]
56. Churchill CH, Abril A, Krishna M, et al. Jaw claudication in primary amyloidosis: unusual presentation of a rare disease. J Rheumatol 2003; 30: 2283. [ Links ]
57. Estrada A, Stenzel TT, Burchette JL, Allen NB. Multiple myeloma-associated amyloidosis and giant cell arteritis. Arthritis Rheum 1998; 41: 1312. [ Links ]
58. Schima W, Amann G, Steiner E, et al. Sicca syndrome due to primary amyloidosis. Br J Radiol 1994; 67: 1023. [ Links ]
59. Jardinet D, Westhovens R, Peeters J. Sicca syndrome as an initial symptom of amyloidosis. Clin Rheumatol 1998; 17: 546. [ Links ]
60. Hachulla E, Janin A, Flipo RM, et al. Labial salivary gland biopsy is a reliable test for the diagnosis of primary and secondary amyloidosis. Arthritis Rheum 1993; 36: 691. [ Links ]
61. Paydas S. Report on 59 patients with renal amyloidosis. Int Urol Nephrol 1999; 31: 619-631. [ Links ]
62. Uda H, Yokota A, Kobayashi K, Miyake T, Fushimi H, Maeda A, et al. Two distinct clinical courses of renal involvement in rheumatoid patients with AA amyloidosis. J Rheumatol 2006; 33: 1482-1487. [ Links ]
63. Kyle RA, Remstein ED, Therneau TM, Dispenzieri A, Kurtin PJ, Hodnefield JM, et al. Clinical course and prognosis of smoldering (asymptomatic) multiple myeloma. N Engl J Med 2007; 356: 2582-2590. [ Links ]
64. Iglesias-Gamarra A, Vasquez-Lamadrid J, Abud-Mendoza C. Enfermedades metabólicas del hueso. Vol. II. Edición INAS, 1992; 545-552. [ Links ]
65. Dispenzieri A. POEMS syndrome. Blood Rev 2007; 21: 285-299. [ Links ]
66. Gejyo F, Odanis S, Yamada T, et al. ß2-microglobulin: A new form of amyloid protein associated with chronic hemodialysis. Kidney Int 1986; 30: 385. [ Links ]
67. Miyata T, Jadoul M, Kurokawa K, et al. ß2-microglobulin in renal disease. J Am Soc Nephrol 1998; 9: 1723. [ Links ]
68. Fenves AZ, Emmett M, White MG, et al. Carpal tunnel syndrome with cystic bone lesions secondary to amyloidosis in chronic hemodialysis patients. Am J Kidney Dis 1986; 7: 130. [ Links ]
69. Floege J, Koch KM. Beta 2-microglobulin associated amyloidosis and therapy with high flux hemodialysis membranes. Clin Nephrol 1994; 42 Suppl 1: s52. [ Links ]
70. Dember LM, Jaber BL. Dialysis-related amyloidosis: late finding or hidden epidemic?. Semin Dial 2006; 19: 105. [ Links ]
71. Jadoul M, Garbar C, Noel H, et al. Histologic prevalence of beta2-microglobulin amyloidosis in hemodialysis: A prospective post-mortem study. Kidney Int 1997; 51: 1928. [ Links ]
72. Schoels M, Jahn B, Hug F, et al. Stimulation of mononuclear cells by contact with cuprophan membranes: Further increase of ß2-microglobulin synthesis by activated late complement components. Am J Kidney Dis 1993; 21: 394. [ Links ]
73. Ben-Chetrit E. Familial Meditteranean Fever (FMF) and renal AA amyloidosis-phenotype-genotype correlations, treatment and prognosis. J Nephrol 2003; 16: 431. [ Links ]
74. Delibas A, Oner A, Balci B, et al. Genetic risk factors of amyloidogenesis in familial Mediterranean fever. Am J Nephrol 2005; 25: 434. [ Links ]
75. Dinc A, Erdem H, Rowczenio D, et al. Autosomal dominant periodic fever with AA amyloidosis: Tumor necrosis factor receptor-associated periodic syndrome (TRAPS) in a Turkish family. J Nephrol 2005; 18: 626. [ Links ]
76. Aganna E, Martinon F, Hawkins PN, et al. Association of mutations in the NALP3/CIAS1/PYPAF1 gene with a broad phenotype including recurrent fever, cold sensitivity, sensorineural deafness, and AA amyloidosis. Arthritis Rheum 2002; 46: 2445. [ Links ]
77. Obici L, Manno C, Muda AO, et al. First report of systemic reactive (AA) amyloidosis in a patient with the hyperimmu-noglobulinemia D with periodic fever syndrome. Arthritis Rheum 2004; 50: 2966. [ Links ]
78. Arostegui JI, Aldea A, Modesto C, et al. Clinical and genetic heterogeneity among Spanish patients with recurrent autoinflammatory syndromes associated with the CIAS1/ PYPAF1/NALP3 gene. Arthritis Rheum 2004; 50: 4045. [ Links ]
79. Ince E, Cakar N, Tekin M, et al. Arthritis in children with familial Mediterranean fever. Rheumatol Int 2002; 21: 213. [ Links ]
80. Balaban B, Yasar E, Ozgul A, et al. Sacroiliitis in familial Mediterranean fever and seronegative spondyloarthropathy: importance of differential diagnosis. Rheumatol Int 2005; 25: 641. [ Links ]
81. Hull KM, Drewe E, Aksentijevich I, et al. The TNF recep-tor-associated periodic syndrome (TRAPS): emerging concepts of an autoinflammatory disorder. Medicine (Baltimore) 2002; 81: 349. [ Links ]
82. Duston MA, Skinner M, Shirahama T, Cohen, AS. Diagnosis of amyloidosis by abdominal fat pad aspiration. Analysis of four years experience. Am J Med 1987; 82: 412. [ Links ]
83. Duston MA, Skinner M, Meenan RF, Cohen AS. Sensitivity, specificity, and predictive value of abdominal fat aspiration for the diagnosis of amyloidosis. Arthritis Rheum 1989; 32: 82. [ Links ]
84. Ansari-Lari MA, Ali SZ. Fine-needle aspiration of abdominal fat pad for amyloid detection: a clinically useful test?. Diagn Cytopathol 2004; 30: 178. [ Links ]
85. van Gameren, Hazenberg BP, Bijzet J, van Rijswijk MH. Diagnostic accuracy of subcutaneous abdominal fat tissue aspiration for detecting systemic amyloidosis and its utility in clinical practice. Arthritis Rheum 2006; 54: 2015. [ Links ]
86. Kyle RA, Bayrd ED. Amyloidosis: review of 236 cases. Medicine (Baltimore) 1975; 54: 271. [ Links ]
87. Cohen AS, Calkins E. Electron microscopic observations on a fibrous component in amyloid of diverse origins. Nature 1959; 183: 1202. [ Links ]
88. Arbustini E, Morbini P, Verga L, Merlini G. Light and electron microscopy immunohistochemical characterization of amyloid deposits. Amyloid 1997; 4: 157. [ Links ]
89. Hawkins PN. Serum amyloid P component scintigraphy for diagnosis and monitoring amyloidosis. Curr Opin Nephrol Hypertens 2002; 11: 649. [ Links ]
90. Hazenberg BP, van Rijswijk MH, Piers DA, et al. Diagnostic performance of 123I-labeled serum amyloid P component scintigraphy in patients with amyloidosis. Am J Med 2006; 119: 355.e15. [ Links ]
91. Mumford AD, ODonnell J, Gillmore JD, et al. Bleeding symptoms and coagulation abnormalities in 337 patients with AL-amyloidosis. Br J Haematol 2000; 110: 454. [ Links ]
92. Choufani EB, Sanchorawala V, Ernst T, et al. Acquired factor X deficiency in patients with amyloid light-chain amyloidosis: Incidence, bleeding manifestations, and response to high-dose chemotherapy. Blood 2001; 97: 1885. [ Links ]
93. Kyle RA, Greipp PR, Garton JP, Gertz MA. Primary systemic amyloidosis. Comparison of melphalan/prednisone versus colchicine. Am J Med 1985; 79: 708-716. [ Links ]
94. Kyle RA, Gertz MA, Greipp PR et al. A trial of three regimens for primary amyloidosis: colchicines alone, melphalan and prednisone, and melphalan, prednisone, and colchicine. N Engl J Med 1997; 336: 1202-1207. [ Links ]
95. Skinner M, Anderson J, Simms R, et al. Treatment of 100 patients with primary amyloidosis: a randomized trial of melphalan, prednisone, and colchicine versus colchicine only. Am J Med 1996; 100: 290-298. [ Links ]
96. Masta A, Gray PJ, Phillips DR. Nitrogen mustard inhibits transcription and translation in a cell free system. Nucleic Acids Res 1995; 23: 3508-3515. [ Links ]
97. Dhodapkar MV, Hussein MA, Rasmussen E, et al. Clinical efficacy of high-dose dexamethasone with maintenance dexamethasone/alpha interferon in patients with primary systemic amyloidosis: results of United States Intergroup Trial Southwest Oncology Group (SWOG) S9628. Blood 2004; 104: 3520-3526. [ Links ]
98. Palladini G, Perfetti V, Obici L, et al. Association of melphalan and high-dose dexamethasone is effective and well tolerated in patients with AL (primary) amyloidosis who are ineligible for stem cell transplantation. Blood 2004; 103: 2936-2938. [ Links ]
99. Dispenzieri A, Lacy MQ, Kyle RA et al. Eligibility for hematopoietic stem-cell transplantation for primarysystemic amyloidosis is a favorable prognostic factor for survival. J Clin Oncol 2001; 19: 3350-3356. [ Links ]
100. Gervais F, Morissette C, Kong X. Proteoglycans and amyloidogenic proteins in peripheral amyloidosis. Curr Med Chem Immun Endocr Metab Agents 2003; 3: 361-370. [ Links ]
101. Dember LM, Hawkins PN, Hazenberg BP, Gorevic PD, Merlini G, Butrimiene I, et al. Eprodisate for AA Amyloidosis Trial Group. Eprodisate for the treatment of renal disease in AA amyloidosis. N Engl J Med 2007; 356: 2349-2360. [ Links ]

