










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Medica Colombiana
Print version ISSN 0120-2448
Acta Med Colomb vol.37 no.4 Bogotá Oct./Dec. 2012
Revisión
Manifestaciones renales de las vasculitis
Renal manifestations of vasculitis
Juan Pablo Camargo1, Jorge de Jesús Cantillo2, Bogotá, D.C. (Colombia)
1Dr. Juan Pablo Camargo Mendoza: Internista, Epidemiólogo Clínico, Hospital Occidente de Kennedy;
2Dr. Jorge de Jesús Cantillo: Profesor Asociado, Departamento de Medicina Interna, Coordinador Sección Nefrología, Facultad de Medicina, Universidad Nacional de Colombia. Docente Clínico, Facultad de Medicina, Universidad de La Sabana y Universidad El Bosque. Nefrólogo, Servicios de Nefrología y Diálisis, Centro Policlínico del Olaya S.A y Hospital Occidente de Kennedy E.S.E. Bogotá D.C. (Colombia).
Correspondencia. Dr. Jorge Cantillo Turbay. Bogotá D.C. (Colombia).
E-mail: md_cantillo@yahoo.com
Recibido: 04/VII/2012 Aceptado: 25/X/2012
Resumen
Objetivo: establecer las principales manifestaciones clínicas, fisiopatológicas, pronóstico y tratamiento de las vasculitis primarias con compromiso renal.
Metodología: se realizó revisión sistemática de la literatura médica consignada en la base de datos Medline, Cochrane y EMBASE con énfasis en trabajos originales y revisiones de tema con impacto en la práctica clínica, en lo referente a vasculitis primarias y riñón, desde 1960 a marzo de 2012. Se procede luego a la consecución de los artículos completos, a su lectura, complementación con artículos referenciados relevantes, y por ultimo al ordenamiento, clasificación y redacción del texto.
Resultados: con los métodos de búsqueda se obtuvieron 1992 resúmenes, de los cuales 83 eran representativos para los tópicos de esta revisión. Al evaluar los artículos completos se consideró que 47 cumplían el objetivo propuesto. Se encontraron descripciones de compromiso renal en todo los tipos de vasculitis. Sin embargo, la mayoría del los estudios con mayor nivel de evidencia se centran en las vasculitis de pequeño vaso asociadas ANCA, principalmente en tratamiento y en factores pronóstico. Las figuras de patología incluidas, son parte de casos recopilados en nuestra práctica clínica.
Conclusión: el compromiso renal de las vasculitis es de mayor impacto en las de pequeño vaso, donde el uso de esteroides e inmunosupresores son pilar fundamental en el tratamiento. Sin embargo hay que resaltar el papel de los agentes biológicos en recaída y procedimientos como la plasmaféresis en casos severos. (Acta Med Colomb 2012; 37: 192-200).
Palabras clave: vasculitis, riñón, glomerulonefritis, autoinmune, ciclofosfamida.
Abstract
Objective: to establish the main clinical manifestations, pathophysiology, prognosis and treatment of primary vasculitis with renal involvement.
Methodology: we performed systematic review of the medical literature contained in the Med-line, Cochrane and EMBASE database with emphasis on original studies and revision of topics with impact on clinical practice referring to primary vasculitis and kidney, from 1960 to March of 2012. We then proceeded in the achievement of the full articles, his reading, complementation with relevant referenced articles, and finally its ordering, clasification and drafting.
Results: with search methods, 1992 abstracts were obtained. 83 were representative for the topics of this review. When evaluating the full papers, we considered that 47 met the objective. Description of renal involvement was found in all types of vasculitis. However, the majority of the studies with the highest level of evidence focus on small vessel vasculitis ANCA associated, mainly on treatment and prognostic factors. Included pathology figures are part of cases collected in our clinical practice.
Conclusion: renal involvement in vasculitis has greater impact on those of small vessel, where the use of steroids and immunosuppressants are the mainstay treatment. However, we must highlight the role of biological agents in case of relapse and procedures such as plasmapheresis in severe cases. (Acta Med Colomb 2012; 37: 192-200).
Keywords: vasculitis, kidney, glomerulonephritis, autoimmune, cyclophosphamide.
Introducción
Las vasculitis son clásicamente definidas como procesos clinicopatológicos caracterizados por inflamación y daño de los vasos sanguíneos (vénulas, capilares, arteriolas de mediano y gran calibre), produciendo las manifestaciones clínicas de acuerdo con la región irrigada, asociándose a necrosis o trombosis, es decir, que las vasculitis pueden ser generalizadas o localizadas. En este contexto se conocen dos categorías: vasculitis secundarias, en la que la inflamación de los vasos ocurre asociada a una enfermedad subyacente o a una exposición y las vasculitis primarias de etiología no muy bien esclarecida en la cual la vasculitis es la causa básica de la injuria tisular (1, 2).
El primer reporte de un paciente con vasculitis no infecciosa se hizo en 1866, cuando Kussmaul y Maier publicaron un informe detallado de un trastorno que se caracteriza por la inflamación nodular de las arterias musculares. Ellos llamaron a esta enfermedad periarteritis nodosa, que más tarde se denominaría poliarteritis nodosa (3). En 1990 el Colegio Americano de Reumatología publicó un estudio con el fin de establecer los criterios para la clasificación de las vasculitis a través de la identificación de las características que las distinguían entre sí. Para el año 1994 se realizó una revisión de la nomenclatura (Clasificación de Chapel-Hill) describiendo 10 formas de vasculitis incluyendo la poliangeítis microscópica (PAM), la enfermedad de Kawasaki y la vasculitis crioglobulinémica. En esta oportunidad se enfatiza la importancia de los ANCA (anticuerpos anticitoplasma del neutrófilo) en el diagnóstico de vasculitis severas al igual que señala los aspectos clínicos, radiológicos, de laboratorio e histológicos para acercarse a una etiología específica. Actualmente se reconocen aproximadamente 20 formas de vasculitis dando reconocimiento a las formas secundarias (Tabla 1) (1, 4-6).

El riñón es afectado con frecuencia en las vasculitis, las de pequeño vaso se asocian a glomerulonefritis, siendo la lesión vasculítica más común y cuya definición histopatológica es glomerulonefritis necrosante. Las vasculitis de mediano vaso ocasionalmente afectan el riñón con una arteritis necrosante y las vasculitis de grande vaso en rara ocasión comprometen el riñón, cuando lo hacen es de manera secundaria por isquemia debida al estrechamiento de las arterias proximales. En este capítulo se describen las vasculitis que afectan el riñón con énfasis en las vasculitis de pequeño vaso particularmente las que están asociadas a ANCA (7).
Material y métodos
Se realizó revisión sistemática de la literatura médica consignada en la base de datos Medline, Cochrane y EMBASE con énfasis en trabajos originales y revisiones de tema con impacto en la práctica clínica, en lo referente a vasculitis primarias y riñón, desde 1960 a marzo de 2012. Utilizando combinación de los siguientes términos MESH: "Vasculitis, Kidney, Anti Neutrophil Cytoplasmic Antibody Associated Vasculitis, Glomerulonephritis, Nephritis, Kidney Failure, Clinical Trial, Review, Systematic Reviews". Se procede luego a la consecución de los artículos completos, a su lectura, complementación con artículos referenciados relevantes, y por ultimo al ordenamiento, clasificación y redacción del texto. Con los métodos de búsqueda se obtuvieron 1992 resúmenes, de los cuales 83 eran representativos para los tópicos de esta revisión. Y al evaluar los artículos completos se considero que 47 cumplían el objetivo propuesto.
Compromiso renal de las vasculitis de vaso grande
Arteritis de células gigantes
Es definida desde el consenso de Chapell Hill como una arteritis de la aorta y de sus ramas principales, con predilección a las ramas extracraneales de las carótidas. Compromete frecuentemente a mayores de 40 años y es relacionada de manera importante con la polimialgia reumática. Proteinuria y hematuria microscópica se ha encontrado en menos de 10% de los pacientes y aun más raro la presencia de insuficiencia renal. Amiloidosis renal secundaria se ha descrito en algunos casos de arteritis de células gigantes y se podría sospechar en pacientes con enfermedad de larga duración que presenten proteinuria y falla renal (8, 9).
Se han descrito síndromes de superposición con este tipo de vasculitis y otras formas con ANCA positivos en el contexto de glomerulonefritis y de igual forma con la poliarteritis nodosa clásica. Por lo que ante sospecha de compromiso renal en poliarteritis de células gigantes es mandatorio incluir ANCA y biopsia renal (10, 11).
Arteritis de Takayasu
Es una inflamación granulomatosa de la aorta y de sus ramas principales, que en contraste con la arteritis de células gigantes es más frecuente en pacientes menores de 40 años con predominio en el sexo femenino. La arteritis en su estado agudo se caracteriza por infiltración de los grandes vasos con células mononucleares, granulocitos y algunas células gigantes. Este proceso inflamatorio lleva a cambios obstructivos por proliferación de la íntima y fibrosis de la media. Aunque los vasos de la parte superior del cuerpo son los más comprometidos, se ha descrito extensión a los vasos abdominales. Compromiso a nivel de las arterias renales puede generar hipertensión renovascular e insuficiencia renal leve debido a la obstrucción de los ostia de las arterias (12). Leve proteinuria y hematuria puede ocurrir, pero enfermedad glomerular o intersticial es infrecuente. Amiloidosis secundaria y nefropatía por Ig A asociada ha sido descrita en pocos casos (13, 14).
La arteritis de Takayasu cuando se encuentra activa se caracteriza por niveles elevados de proteína C reactiva, en este caso los esteroides son fundamentales en algunos casos con asociación de inmunosupresores. Estenosis de la arteria renal puede ser tratada con radiología intervencionista, como la angioplastia renal transluminal con o sin colocación de stent y en su defecto cirugía reconstructiva (15).
Compromiso renal de las vasculitis de medianos vasos
Enfermedad de Kawasaki
Es una vasculitis poco frecuente que afecta niños en todas las partes del mundo y representa la primera causa de enfermedad cardiaca en Estados Unidos y Japón (16). El compromiso primario del riñón es raro, pero se ha reportado falla renal aguda de tipo prerrenal por disminución del volumen intravascular efectivo y compromiso en el gasto cardiaco; por otra parte se han reportado algunos casos de nefritis. La ecografía renal ha mostrado en estos casos aumento del tamaño con incremento de la ecogenecidad. Algunos reportes de casos muestran en la histopatología cambios de enfermedad de complejos inmunes, nefritis intersticial y esclerosis mesangial (17-20).
Poliarteritis nodosa clásica
Este tipo de vasculitis se caracteriza por inflamación necrosante de arterias de mediano y pequeño vaso. Glomerulonefritis o vasculitis en arteriolas, capilares o vénulas es ausente. Por otra parte los ANCA son generalmente negativos. En la histopatología se describe abundantes neutrófilos, cambios fibrinoides y disrupción de la lámina elástica interna. Los infartos corticales descritos son resultado de la isquemia por obliteración aguda o crónica. En las aéreas de necrosis, los aneurismas pueden aparecer con ruptura resultando en hemorragia retroperitoneal y consecuente falla prerrenal (2, 21).
La hipertensión de origen renovascular está presente en 50% de los pacientes, se describen síntomas como dolor en flanco y hematuria relacionados con ruptura de microaneurismas. La biopsia renal muestra cambios característicos de esta vasculitis en las arterias renales, no obstante debido al riesgo de hemorragia por ruptura de aneurismas este procedimiento es generalmente evitado. La angiografía renal puede detectar aneurismas, otros defectos no aneurismáticos, arterias colaterales y retraso de vaciamiento de las arterias renales. En los pacientes con compromiso agudo se considera esquema de tratamiento con glucocorticoides y ciclofosfamida. De igual forma el tratamiento antihipertensivo debe buscar las metas para evitar la progresión de la falla renal. Las hemorragias mayores podrían ser tratadas con técnicas de radiología intervencionista con embolización (22).
Compromiso renal de las vasculitis de pequeño vaso con ANCA positivo
Granulomatosis de Wegener (granulomatosis con poliangeítis)
Esta vasculitits cuyo nombre puede ser cambiado a granulomatosis con poliangeítis, en vista del pasado Nazi del doctor Friedrich Wegener (23). Es un desorden autoinmune de etiología no establecida, pero donde la infección podría esclarecer esta pregunta, caracterizado por inflamación granulomatosa que involucra el tracto respiratorio y causa vasculitis necrosante de pequeño y mediano vaso. La tasa de incidencia es de 6 a 12 por millón/año en el Reino Unido, Alemania y Noruega. Bajas incidencias se encuentran en el sur de Europa, países como España reportan tasas de 2.9 por millón/año. En Japón la incidencia es similar, sin embargo, la presentación clínica es diferente ya que la progresión a falla renal es rara, lo que orienta a un fenotipo especial (24).
Proteinasa 3 (PR3)-ANCA están positivos en el 90 a 95% de los casos de la granulomatosis de Wegener (WG). La serina proteinasa 3 (PR3) es el mayor autoantígeno de la WG y es almacenado en los gránulos azurófilos de los neutrófilos. La expresión de esta proteína se relaciona con la actividad de la enfermedad y el riesgo de recaída. De forma inicial las citoquinas proinflamatorias de los neutrófilos como FNT (factor de necrosis tumoral)-a aumentan la expresión de PR3 en la membrana externa de los mismos. Translocación de PR3 de los gránulos azurófilos a la superficie de los neutrófilos es disponible para la unión PR3-ANCA. La unión de PR3-ANCA tanto a PR3 y a receptores Fcy es lo que perpetúa la actividad de los neutrófilos, a través de señales del sistema de transducción por la vía FcyRIIa. El FNT-a liberado por los neutrófilos y activado por PR3-ANCA induce alteración de la cadena respiratoria y degranulación, resultando en la liberación de citocinas proinflamatorias, proteasas y reactantes de oxígeno, que conllevan a daño vascular. In vitro los estudios han mostrado que FNT-a regula la adhesión de moléculas en las células endoteliales, PR3-ANCA activado es capaz de adherirse al endotelio activado y disparar la citotoxicidad hacia las células endoteliales. La capacidad para que los neutrófilos se activen en respuesta a la expresión de citoquinas y PR3-ANCA, y la capacidad de las citocinas tales como FNT-a para regular la expresión de moléculas de adhesión ha llevado a la comprensión del papel patogénico de PR3-ANCA en vasculitis de pequeños vasos y la comprensión de la interacción entre PR3-ANCA, los neutrófilos, las citoquinas (FNT-a) y el endotelio (25).
En cuanto a la presentación clínica inicial 38 a 70% de los pacientes presentan signos de compromiso renal dado por proteinuria, anormalidades del sedimento urinario y disminución de la función renal. Más de 50% de los pacientes que se presentan sin compromiso renal en un inicio, lo manifiestan en el curso de la enfermedad. La función renal inicial es un buen predictor pronóstico, aproximadamente 10-20% de los pacientes desarrollan falla renal con requerimiento de hemodiálisis concomitante a la terapia inmunosupresora. Lo anterior da manifestaciones clínicas variables, típicamente la presentación de los pacientes con GW consiste en un periodo insidioso de síntomas de la vía aérea superior, debilidad generalizada, mialgias y artralgias. El paso a glomerulonefritis rápidamente progresiva con rápida declinación de la función renal asociado a otros signos de glomerulonefritis es variable (proteinuria que no progresa a síndrome nefrótico) y hematuria con cilindros celulares (26-29).
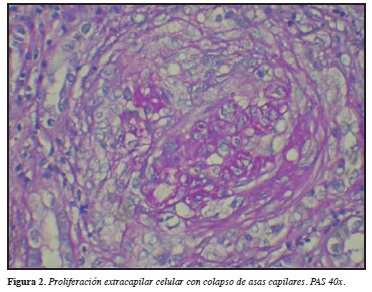
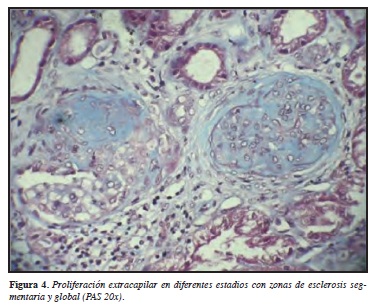
Histopatológicamente, la fase inicial es caracterizada por inflamación endotelial con infiltración de granulocitos de polimorfonucleares con posterior necrosis fibrinoide de la pared de los capilares y trombosis intracapilar. Posterior a esto desarrollan células en media luna a lo largo de la extensión del proceso vasculítico. Las anteriores células llevan a la destrucción de la cápsula de Bowman, y concomitante a esto se genera inflamación periglomerular, en donde no es infrecuente la formación de pseudogranuloma. El intersticio es involucrado en la mayoría de los casos con infiltración de linfocitos, monocitos, células plasmáticas y leucocitos polimorfonucleares. Formación de granuloma ocurre particularmente en el intersticio en cerca de 10% de los casos. Vasculitis necrosante se observa en algunas ocasiones en las pequeñas arterias intersticiales y en estados avanzados de esclerosis. Células en media luna llevan a fibrosis global o a glomeruloesclerosis difusa (Figuras 1-3). Es importante aclarar que lo estudios de inmunofluorescencia no han demostrado de manera clara depósitos inmunes a lo largo de la pared de los capilares glomerulares, algunos depósitos de IgM y C3 se han visto particularmente en el espacio de Bowman. Basado en estos hallazgos la histología renal se ha descrito como una glomerulonefritis pauci-inmune. Sin embargo, por microscopía electrónica depósitos de complejos inmunes son encontrados en la mitad de las biopsias renales, lo que sugiere un papel importante en el proceso de la enfermedad (30).


Antes de considerar la biopsia renal debe valorarse el grado de actividad de la enfermedad. Los exámenes que deben incluirse son la creatinina sérica, depuración de creatinina en recolección de orina de 24 horas, uroanálisis con valoración de sedimento e identificar cilindros. La biopsia renal podría ser considerada en el caso de que no hubiese un diagnostico claro y si el compromiso renal es significativo (31).
La GW en 1995 era considerada una enfermedad granu-lomatosa que progresaba a vasculitis generalizada, determinando dos estadios. En las actuales definiciones (Tabla 2), el estadio localizado es restringido a manifestaciones del aérea superior o inferior sin signos de vasculitis sistémica, se diferencia de los estados sistémicos (temprano, generalizado y enfermedad severa). El estadio temprano no compromete la vida, el generalizado lleva a disfunción de órgano y en el estadio severo ya existe falla de órgano en el contexto de esta revisión (creatinina > 5.7 mg/dL). Pacientes mayores de 50 años con compromiso de la función renal sin alteración en nariz, orejas o garganta son relacionados con mal pronóstico. Con lo nombrado anteriormente se dan las recomendaciones del EULAR para tratamiento de vasculitis de pequeño vaso (Tabla 3) que se considera como guía para esta revisión, complementado con los resultados del RAVE-ITN en cuanto superioridad de rituximab comparado con manejo convencional en recaídas (32, 35).


Poliangeítis microscópica
Es una enfermedad autoinmune caracterizada por vasculitis sistémica que compromete vasos de pequeño calibre, conllevando a capilaritis pulmonar y glomerulonefritis, siendo la primera causa de síndrome pulmón-riñón, de un grupo de patologías que incluyen el síndrome de Goodpasture, el lupus eritematoso sistémico y la GW (33).
La PAM típicamente se presenta en mayores de 50 años y la mayoría de series reporta predominio en hombres. La PAM es más prevalente que la GW en poblaciones asiáticas, pero su prevalencia es más baja en países del occidente, un estudio sueco mostró incidencias similares entre ambas (10/1 millón de habitantes por año). Otros datos muestran incremento en la incidencia de la PAM con la edad: por otra parte se ha mostrado influencia de la latitud geográfica y la exposición de los rayos ultravioleta para la WG y SCS pero aún no se ha verificado en la PAM (34).
Los ANCA desempeñan un papel fundamental en la patogénesis de la PAM. En teoría puede ocurrir en dos partes. La primera en donde los neutrófilos son expuestos a dosis bajas de citocinas proinflamatorias como IL-1 (interleuquina 1) o FNT-a. Este proceso lleva a la expresión de MPO (mieloperoxidasa), seguido de la adherencia de neutrófilos a la superficie del endotelio de los vasos sanguíneos y el glomérulo. En la segunda parte es donde los neutrófilos son activados por interacción con ANCA-MPO, con unión a sus sustratos o se integra con los receptores Fc de los neutrófilos. Los modelos animales han demostrado que la unión ANCA-MPO es suficiente para inducir capilaritis y glomerulonefritis, no obstante en los humanos su presentación puede variar, lo que orienta a que pueden existir otros cofactores incluyendo la predisposición genética (33).
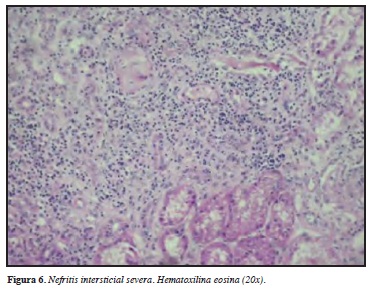
El compromiso renal se caracteriza por una glomerulonefritis rápidamente progresiva, siendo la principal característica clínica de la PAM. El 80-100% de los pacientes experimentan manifestaciones renales, la cual va de un rango de sedimento urinario positivo en paciente asintomático hasta enfermedad renal terminal requiriendo diálisis. Durante la glomerulonefritis la proteinuria es el hallazgo primordial (rango nefrótico en 50% de los pacientes), seguido de hematuria microscópica y gránulos eritrocitarios en el sedimento. El principal hallazgo en la biopsia renal es glomerulonefritis segmentaria necrosante en 98% de los pacientes con compromiso renal (Figuras 4-6). Células en media luna glomerulares son también comunes, y se han documentado hasta 90% de los casos. Vasculitis franca y necrosis fibrinoide se observa tan sólo en 20% de los pacientes. Otras formas de compromiso glomerular se han visto; por ejemplo nefritis intersticial y atrofia tubular en 20% de los casos. La inmunofluorescencia muestra mínimos depósitos de inmunoglobulinas o complemento en los vasos renales y el glomérulo (34).

En cuanto el tratamiento, la mayoría de los estudios prospectivos que incluyen GW también incluyen PAM, por características fisiopatológicas similares. No obstante las presentaciones severas como el síndrome pulmón-riñón es más frecuente en la PAM, por lo que un régimen de tratamiento más agresivo es prescrito. Como primera línea de tratamiento se encuentran los esteroides y los citotóxicos usualmente la de primera elección es la ciclofosfamida. Recientes reuniones de consenso han demostrado igual eficacia entre pulsos de ciclofosfamida endovenosa en comparación a la oral. En cuanto al metotrexate no es recomendado por toxicidad tubular. Para el mantenimiento de la remisión se recomienda micofenolato de mofetilo o azatriopina sin ninguna ventaja en el primero en cuanto recaída, sin embargo es usado en casos de intolerancia o efectos secundarios de la azatriopina. La plasmaféresis es una opción en glomerulonefritis responsable de falla renal, el grupo EUVAS (European Vasculitis Study Group) ha demostrado que este procedimiento es superior a los esteroides en la mejoría de la función renal pero no hubo impacto en la sobrevida. En cuanto los agentes biológicos inducen igualmente remisión, sin embargo, su superioridad al tratamiento convencional usando glucocorticoides y ciclofosfamida no ha sido documentada, como lo corrobora el trabajo del grupo de investigación RAVE-ITN, sin embargo hay que destacar su superioridad en el caso de recaídas (35).
Síndrome de Churg-Strauss
Es definido como una inflamación granulomatosa rica en eosinófilos, que compromete el tracto respiratorio y que presenta vasculitis necrosante de pequeños y medianos vasos asociándose con asma y eosinofilia. La incidencia de este desorden se encuentra entre 0.11-2.66 casos por millón de habitantes y tiene una prevalencia de 10.7-14 por millón de habitantes. La relación de hombre a mujer es de 0.3-2.3, con un promedio de presentación que oscila entre 38-52 años. Las lesiones vasculares y glomerulares pueden ser similares a la PAM y GW, aunque típicamente hay un infiltrado de eosinófilos más prominente, un hallazgo característico pero no exclusivo de SCS (36).
Estudios recientes han encontrado ANCA positivo usualmente p-ANCA en 38% de los pacientes. Sin embargo en análisis de subgrupos encontró que la positividad de los ANCA era más probable con manifestaciones de pequeños vasos, incluyendo glomerulonefritis necrosante, mononeuritis y púrpura, y los casos de ANCA negativo en compromiso cardiaco y pulmonar. Lo anterior ha orientado hacia dos mecanismos patogénicos: un proceso mediado por ANCA similar a la GW y PAM y otro proceso de infiltración de eosinófilos con la liberación de productos tóxicos, lo que da la teoría de procesos genéticos distintos (37).
La mayoría de los pacientes presenta alteración renal leve, siendo raro el compromiso severo. Se describe alteración en 50% de los pacientes. La presentación clínica no difiere de la PAM y GW: hematuria microscópica con cilindros celulares, proteinuria y grados variables de insuficiencia renal. A nivel histológico se encuentra infiltración de eosinófilos activos, particularmente en el intersticio, acompañado de formación de granulomas. En la mayoría de los casos se observa glomerulonefritis crecéntica y necrosante focal o difusa. En la inmunofluorescencia directa los depósitos inmunes son generalmente ausentes, lo que la clasifica como una glomerulonefritis pauci-inmune (38, 39).
Los corticoides son el pilar en el tratamiento inicial mejorando dramáticamente el pronóstico. De igual forma algunos pacientes requieren mantenimiento, para lo cual el French Vasculitis Study Group ha definido cinco factores pronóstico en vasculitis necrosante incluyendo SCS: 1) creatinina > 1.58 mg/dL; 2) proteinuria > 1 g/d; 3) compromiso gastrointestinal; 4) cardiomiopatía; 5) compromiso de sistema nervioso central.
En pacientes sin factores de mal pronóstico, es decir, sin ninguno de los anteriormente nombrados, la remisión clínica puede ser obtenida en 93% de los pacientes sólo con esteroide. No obstante, 35% puede presentar recaída en el primer año, por lo que es requerido tratamiento adicional con inmunosupresor ya sea azatriopina o ciclofosfamida. En pacientes con factores de mal pronóstico pulsos de ciclofosfamida asociado a esteroides han mostrado el mejor control de la enfermedad severa, no obstante existen aún discrepancias estadísticas (40).
Púrpura de Henoch Schonlein
Esta vasculitis es más común en la niñez, con una incidencia de 10-20 casos por 100.000 niños/año. Generalmente es autolimitada con un curso promedio de cuatro semanas. Se conoce asociación de esta vasculitis con una variedad de patógenos microbianos y otros agentes medioambientales. Es una vasculitis caracterizada por depósitos inmunes de IgA que afectan pequeños vasos. El compromiso renal se ha reportado en 33% de niños y 63% de adultos. La enfermedad es usualmente leve y sin secuelas serias. Sin embargo, una minoría de los pacientes adultos puede progresar a enfermedad renal terminal. Algunos estudios han demostrado depósitos de IgA mesangiales granulares, frecuentemente en combinación con complemento C3 y fibrinógeno. Los depósitos de IgA son de predominio de la clase IgA1. Los grados de injuria renal varían: en algunos casos hay mínima anormalidad en la microscopía de luz. Proliferación mesangial sin proliferación endocapilar y formación crecente está presente en 25% de las biopsias. En 75% restante existe una extensa proliferación mesangial con proliferación focal a difusa endocapilar acompañado de glomerulonefritis crecéntica y membranoproliferativa (Figuras 7, 8). Los resultados del examen histopatológico son importantes porque determinan el grado de severidad y determina pronóstico. Grandes proporciones de glomérulos comprometidos se relaciona con un incremento en el riesgo de desarrollar falla renal terminal (41).


El valor del uso de esteroides tempranamente como prevención de la nefritis ha sido debatido por muchos años. Varios trabajos como el de Dudley han mostrado que el tratamiento temprano no es efectivo en la prevención de nefritis (42-44).
En cuanto el manejo de la nefritis establecida la prednisona acelera la resolución en los casos leves (hematuria y proteinurias mínimas). Vale la pena señalar, que alteraciones renales menores se resuelven espontáneamente en la mayoría de los pacientes, independientemente del tratamiento. No hay evidencia que los corticosteroides como monoterapia tengan beneficio en el tratamiento de pacientes con nefritis severa asociada a proteinuria en rango nefrótico, sin embargo, los estudios son de bajo poder estadístico, tampoco le dan beneficio significativo a los inmunosupresores. Faltan ensayos clínicos aleatorizados (41).
Vasculitis crioglobulinémica renal
La crioglobulinemia es definida por la presencia de inmunoglobulinas circulantes que se precipitan a temperaturas menores de 37 °C y se disuelven con el recalentamiento. De acuerdo con la composición del crioprecipitado las vasculitis se clasifican en tres subtipos serológicos: crioinmunoglobulinemia monoclonal (tipo I) compuesta por inmunoglobulina monoclonal, crioglobulinemia mixta (tipo II) compuesta por IgG mono y policlonal y la crioglobulinemia policlonal (tipo III) con factor rematoideo de IgM. Muchos casos de las tipo II y III son asociadas a infección por el virus de la hepatitis C. Este tipo de vasculitis son un prototipo de vasculitis mediadas por inmunocomplejos en el cual las crioglobulinas son demostradas en sangre, niveles de C3 y C4 son disminuidos y los depósitos inmunes están presentes en la pared de los vasos (45).
Los tres tipos de vasculitis pueden llevar a compromiso renal, la tipo II en 75% de los casos, la tipo III hasta en 25% y la tipo I en menos de 5%. La glomerulonefritis membranoproliferativa tipo I es el hallazgo típico. Hay que tener en cuenta que 10-30% de los pacientes con hepatitis B crónica desarrollan este tipo de compromiso. Los pacientes con crioglobulinemia y glomerulonefritis membranoproliferativa desarrollan proteinuria (41%), síndrome nefrótico (21%), síndrome nefrítico (21%), falla renal crónica (12%) y falla renal aguda (9%). Síntomas extrarrenales como el compromiso en la piel puede estar ausente en 30-80% de los casos.
En la histología se evidencian depósitos subendoteliales de complejos inmunes de crioglobulinemia y frecuentemente complemento C3 en la membrana basal glomerular. Subsecuente existe proliferación celular mesangial, con marcado engrosamiento de la membrana basal e hipercelularidad glomerular (46).
La crioglobulinemias asintomáticas no requieren tratamiento. El manejo de las secundarias está dirigido a la enfermedad primaria. En cuanto la crioglobulinemia tipo II asociada a hepatitis, las estrategias antivirales son superiores al manejo inmunosupresor. En el resto de tipos los esteroides, inmunosupresores y plasmaféresis son utilizados dependiendo de la severidad de la enfermedad (47).
Conclusiones
El compromiso renal de las vasculitis es de mayor impacto en las de pequeño vaso, donde el uso de esteroides e inmunosupresores son pilar fundamental en el tratamiento. Sin embargo, hay que resaltar el papel de los agentes biológicos en recaída y procedimientos como la plasmaféresis en casos severos.
Es para destacar los posibles cambios en cuanto la clasificación de las vasculitis, en vista de nuevos conceptos fisiopatológicos, que en el caso del síndrome de Goodpasture, la descartaría. Y por otra parte el cambio de nombre y eliminación de epónimos como en la granulomatosis de Wegener.
Referencias
1. Saleh A, Stone JH. Classification and diagnostic criteria in systemic vasculitis. Best Pract Res Clin Rheumatol 2005; 19: 209-21. [ Links ]
2.Langford CA. Vasculitis. J Allergy Clin Immunol 2010; 125: 216-25. [ Links ]
3. Kussmaul A, Maier R. Ueber eine bisher nicht beschriebene eigenthümliche Arterienerkrankung (Periarteritis nodosa), die mit Morbus Brightii und rapid fortschreitender allgemeiner Muskella¨hmung einhergeht. Dtsch Arch Klin Med 1866; 1: 484-518. [ Links ]
4. Hunder GG, Arend WP, Bloch DA, Calabrese LH, Fauci AS, Fries JF, et al. The American College of Rheumatology 1990 criteria for the classification of vasculitis: introduction. Arthritis Rheum 1990; 33: 1065-1067. [ Links ]
5. Bloch DA, Michel BA, Hunder GG, McShane DJ, Arend WP, Calabrese LH, et al. The American College of Rheumatology 1990 criteria for the classification of vasculitis: patients and methods. Arthritis Rheum 1990; 33: 1068-1073. [ Links ]
6. Jennette JC, Falk RJ, Andrassy K, Bacon PA, Churg J, Gross WL, et al. Nomenclature of systemic vasculitides: proposal of an international consensus conference. Arthritis Rheum 1994; 37: 187-192. [ Links ]
7. John R, Herzenberg AM. Vasculitis affecting the kidney. Semin Diagn Pathol 2009; 262: 89-102. [ Links ]
8. Klein RG, Hunder GC, Stanson AW, Sheps SC. Large artery involvement in giant cell (temporal) arteritis. Ann Intern Med 1975; 83: 806-812. [ Links ]
9. Escriba A, Morales E, Albizua E, Herrero JC, Ortuno T, Carreno A, et al. Secondary amyloidosis in patients with polymyalgia rheumatica. Am J Kidney Dis 2000; 35: 137-140. [ Links ]
10. Muller E, Schneider W, Kettritz U, Schmidt WA , Luft FC, Gobel U. Temporal arteritis with pauci-inmune glomerulonephritis: a sytemic disease. Clin Nephro 2000; 62: 384-386. [ Links ]
11. O' Neill WM, Hammar SA, Bloomer HA. Giant cell arteritis with visceral angiitis. Arch Intern Med 1976; 136: 1157. [ Links ]
12. Lagneau P, Michael JB. Renovascular hypertension and Takayasu's disease. J Urol 1985: 134-876. [ Links ]
13. Wada Y, Nishida H, Kohno K, Tamai O, Fujisawa M, Katoh S, et al. A.A amyloidosis in Takayasu's arteritis long term survival on maintenance haemo-dialysis. Nephrol Dial Transplant 1999; 14: 2478-2481. [ Links ]
14. Cavatorta F, Campisi S, Trabassi E, Zollo A, Salvidio G. IgA nephropathy associated with Takayasu's arteritis report of a case and review of the literature. Am J Nephrol 1995; 15: 165-167. [ Links ]
15. Lovaria A, Nicolini A, Meregagia D, Saccheri S, Rivolta R, Montanari E, et al. Interventional radiology in the treatment of renal artery stenosis. Ann Urol 1999; 33: 146-155. [ Links ]
16. Khasnis A, Langford CA. Update on vasculitis. J Allergy Clin Immunol 2009; 123: 1226-36. [ Links ]
17. Senzaki H, Suda M, Noma S, Kawaguchi H, Sakakihara Y, Hishi T. Acute heart failure and acute renal failure in Kawasaki disease. Acta Paediatr Jpn 1994; 36: 443-7. [ Links ]
18. Salcedo JR, Greenberg L, Kapur S. Renal histology of mucocutaneous lymph node syndrome (Kawasaki disease). Clin Nephrol 1998; 29: 47-51. [ Links ]
19. Veiga PA, Pieroni D, Baier W, Feld LG. Association of Kawasaki disease and acute interstitial nephritis. Pediatr Nephrol 1992; 6: 421-423. [ Links ]
20. Joh K, Kanetsuma Y, Ishikawa Y, Aizawa S, NAito L, Sado Y. Diffuse mesangial sclerosis associated with Kawasaki disease: analysis of alpha chains (alpha 1-alpha 6) of human type 1 in the renal basement membrane. Virchows Archiv 1997; 430: 489-94. [ Links ]
21. Chandrakantan A, Kaufman J. Renal hemorrhage in polyarteritis nodosa: diagnosis and management. Am J Kidney Dis 1999; 33(6): 8. [ Links ]
22. Brogan P A, Davies R, Gordon I, Dillon M J. Renal angiography in children with PAN. Pediatr Nephrol 2002; 17: 277-283. [ Links ]
23. Falk RJ, Gross WL, Guillevin L, Hoffman GS, Jayne DR, Jennette JC, et al. Granulomatosis With Polyangiitis (Wegener's): An Alternative Name for Wegener's Granulomatosis. Arthritis Rheum 2011; 63: 863-864. [ Links ]
24. Holle JU, Laudien M, Gross WL. Clinical manifestations and treatment of Wegener's granulomatosis. Rheum Dis Clin North Am 2010; 36: 507-26. [ Links ]
25. Schilder AM. Wegener's Granulomatosis vasculitis and granuloma. Autoimmun Rev 2010; 9: 483-7. [ Links ]
26. Reinhold-Keller E, Beuge N, Latza U, de Groot K, Rudert H, Nölle B, et al. An interdisciplinary approach to the care of patients with Wegener's granulomatosis: long-term outcome in 155 patients. Arthritis Rheum 2000; 43: 1021-32. [ Links ]
27. Anderson G, Coles ET, Crane M, Douglas AC, Gibbs AR, Geddes DM, et al. Wegener's granuloma. A series of 265 British cases seen between 1975 and 1985. A report by a sub-committee of the British Thoracic Society Research Committee. QJ Med 1992; 83: 427-38. [ Links ]
28. Hoffman GS, Kerr GS, Leavitt RY, Hallahan CW, Lebovics RS, Travis WD, Rottem M, Fauci AS. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med 1992; 116: 488-98. [ Links ]
29. Abdou NI, Kullman GJ, Hoffman GS, Sharp GC, Specks U, McDonald T, Garrity J, Goeken JA, Allen NB. Wegener's granulomatosis: survey of 701 patients in North America. Changes in outcome in the 1990s. J Rheumatol 2002; 29: 309-16. [ Links ]
30. Hass M, Eustace JA. Inmune complex deposits in ANCA associated rescentic glomerulonephritis: A study of 126 cases. Kidney Int 2004; 65: 2145-2147. [ Links ]
31. Booth AD, Almond MK, Burns A, Ellis P, Gaskin G, Neild GH, et al. Outcome of ANCA associated renal vasculitis: a 5 year retrospective study. Am J Kidney Dis 2003: 41: 777-783. [ Links ]
32. Mukhtyar C, Guillevin L, Cid MC, Dasgupta B, de Groot K, Gross W, et al. EULAR recommendations for the management of primary small and medium vessel vasculitis. Ann Rheum Dis 2009; 68: 310-7. [ Links ]
33. Chung SA, Seo P. Microscopic Polyangiitis. Rheum Dis Clin N Am 2010; 36: 545-558. [ Links ]
34. Villiger PM, Guillevin L. Microscopic polyangiitis: Clinical presentation. Autoimmun Rev 2010; 9: 812-9. [ Links ]
35. Stone JH, Merkel PA, Spiera R, Seo P, Langford CA, Hoffman GS, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med 2010; 363: 221-32. [ Links ]
36. Ntatsaki E, Watts RA, Scott DG. Epidemiology of ANCA-associated vasculitis. Rheum Dis Clin North Am 2010; 36: 447-61. [ Links ]
37. Baldini C, Talarico R, Della Rossa A, Bombardieri S. Clinical manifestations and treatment of Churg-Strauss syndrome. Rheum Dis Clin North Am 2010; 36: 527-43. [ Links ]
38. Gaskin G, Clutterbuck EJ, Pusey CD. Renal disease in the Churg-Strauss syndrome. Diagnosis, management and outcome. Contrib Nephrol 1991; 94: 58-65. [ Links ]
39. Sinico RA, Bottero P. Churg-Strauss angiitis. Best Pract Res Clin Rheumatol 2009; 23: 355-6. [ Links ]
40. Ribi C, Cohen P, Pagnoux C, Mahr A, Arène JP, Lauque D, et al. Treatment of Churg-Strauss syndrome without poor-prognosis factors: a multicenter, prospective, randomized open-label study of seventy-two patients. Arthritis Rheum 2008; 58: 586-94. [ Links ]
41. Saulsbury FT. Henoch-Schönlein purpura. Curr Opin Rheumatol 2010; 22: 598-602. [ Links ]
42. Narchi H. Risk of long term renal impairment and duration of follow up recommended for Henoch-Schonlein purpura with normal or minimal urinary findings: a systematic review. Arch Dis Child 2005; 90: 916-920. [ Links ]
43. Wakaki H, Ishikura K. Henoch-Schönlein nephritis with nephrotic state in children: predictors of poor outcomes. Pediatr Nephrol 2012; 27: 335. [ Links ]
44. Soylemezoglu O, Ozkaya O, Ozen S, Bakkaloglu A, Dusunsel R, Peru H et al. Henoch-Schonlein nephritis: a nationwide study. Nephron Clin Pract 2009; 112: 199-204. [ Links ]
45. Ferri C, Mascia MT. Cryoglobulinemic vasculitis. Curr Opin Rheumatol 2006 18: 54-63. [ Links ]
46. Braun GS, Horster S, Wagner KS, Ihrler S, Schmid H. Cryoglobulinaemic vasculitis: classification and clinical and therapeutic aspects. Postgrad Med J 2007; 83: 87-94. [ Links ]
47. Ferri C. Mixed cryoglobulinemia. Orphanet J Rare Dis 2008; 16: 25. [ Links ]
