










Services on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Medica Colombiana
Print version ISSN 0120-2448
Acta Med Colomb vol.46 no.4 Bogotá Oct./Dec. 2021 Epub May 02, 2022
https://doi.org/10.36104/amc.2021.2082
Presentación de casos
Miocardiopatía hipertrófica apical. Síndrome de Yamaguchi
a Residente de Medicina Interna Universidad Libre; Cali (Colombia).
b Internista, Cardiólogo. Profesor Asistente Departamento de Medicina Interna, Universidad del Valle, DIME Clínica Neurocardiovascular; Cali (Colombia).
c Internista, Cardiólogo. Profesor Asistente Departamento de Medicina Interna, Universidad del Valle, DIME Clínica Neurocardiovascular. Cardiólogo Electrofisiólogo Clínica Imbanaco; Cali (Colombia).
d Médica General Clínica Imbanaco. Cali (Colombia).
Introducción:
la miocardiopatía hipertrófica (MCH) apical constituye 15% de los pacientes con MCH en Japón; sin embargo, en nuestro país es un diagnóstico poco frecuente. Esta entidad consiste en la afectación casi exclusiva del ápex, siendo más común su localización ventricular izquierda. Su diagnóstico se da por la visualización de engrasamiento apical mayor a 15 mm o una relación entre el espesor de la pared apical y basal del ventrículo izquierdo >1.3-1.5. Su tratamiento es sintomático a través del uso de betabloqueadores y el implante de desfibrilador automático para prevención primaria de muerte súbita.
Presentación de caso:
paciente de 59 años que de forma incidental se le confirmó MCH apical o síndrome de Yamaguchi.
Conclusión:
la MCH apical es una entidad infrecuente, en muchos casos asintomática. Su diagnóstico oportuno permite un tratamiento temprano y prevenir desenlaces cardiovasculares incluso fatales. (Acta Med Colomb 2021; 46. DOI:https://doi.org/10.36104/amc.2021.2082).
Palabras clave: cardiomiopatía hipertrófica; electrocardiografía
Introduction:
apical hypertrophic cardiomyopathy (HCM) accounts for 15% of patients with HCM in Japan. However, in our country it is a rare diagnosis. In this entity, the apex is almost exclusively affected, most commonly in the left ventricle. It is diagnosed by finding apical thickening greater than 15 mm or a ratio of left ventricular apical to basal wall thickness >1.3-1.5. It is treated symptomatically with beta blockers and automatic defibrillator implantation for primary prevention of sudden death.
Case presentation:
a 59-year-old patient in whom apical HCM, or Yamaguchi syndrome, was found incidentally.
Conclusion:
apical HCM is a rare entity, in many cases asymptomatic. Its prompt diagnosis allows early treatment and prevents cardiovascular outcomes, including fatal ones. (Acta Med Colomb 2021; 46. DOI:https://doi.org/10.36104/amc.2021.2082).
Keywords: hypertrophic cardiomyopathy; electrocardiography
Introducción
La miocardiopatía hipertrófica es la causa más común de miocardiopatía hereditaria, que afecta una de cada 500 personas 1. La miocardiopatía hipertrófica puede comprometer todo el corazón o por separado los segmentos basales, los segmentos medioventriculares y apicales de ambos ventrículos. La variedad apical es rara, usualmente involucra el ventrículo izquierdo y raramente el ventrículo derecho o ambos 2. Fue descrita inicialmente en Japón en 1976 y se caracteriza por una cavidad ventricular izquierda con configuración en "As de picas" al final de la diástole en la ventriculografía 3. Históricamente se pensaba que esta condición estaba confinada a la población japonesa, que es donde se encuentra su mayor prevalencia. De todos los pacientes con miocardiopatía hipertrófica en Japón, la prevalencia de la variante apical fue 15%, mientras que en Estados Unidos es 3% 2.
Su presentación clínica es heterogénea y el hallazgo electrocardiográfico de negatividad de una onda T gigante ha sido reportada como la manifestación típica de esta enfermedad. Su diagnóstico imagenológico por medio de ecocardiografía puede tener limitaciones en la evaluación del ápex, por lo cual, la resonancia magnética (RM) cardiaca, se ha consolidado como un método alternativo no invasivo de elección desplazando a la ventrículografía que se consideraba el método de oro para su diagnóstico 4. El objetivo de este reporte de caso, es describir la presencia de una variante rara de una enfermedad genética y con potencial morbimortalidad cardiovascular.
Reporte de caso
Paciente varón de 59 años, sin antecedentes patológicos previos ni familiares de muerte súbita o enfermedad cardiovascular manifiesta, quien incidentalmente (consulta al caer accidentalmente de una silla con trauma en tórax en tejidos blandos) fue evaluado clínica y electrocardiográficamente. Ocasionalmente manifestaba palpitaciones de corta duración sin síncope, clase funcional I de NYHA y sin síntomas de enfermedad cardiaca.
El electrocardiograma en reposo revelaba alteración de la repolarización ventricular, con T negativa profunda en V2 y V3, con QT corregido normal e hipertrofia ventricular izquierda (Figuras 1 y 2).
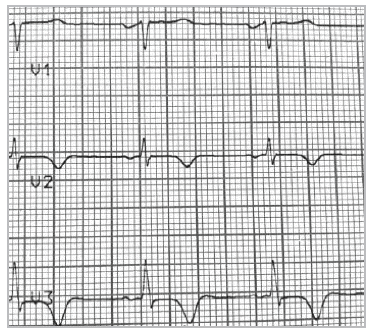
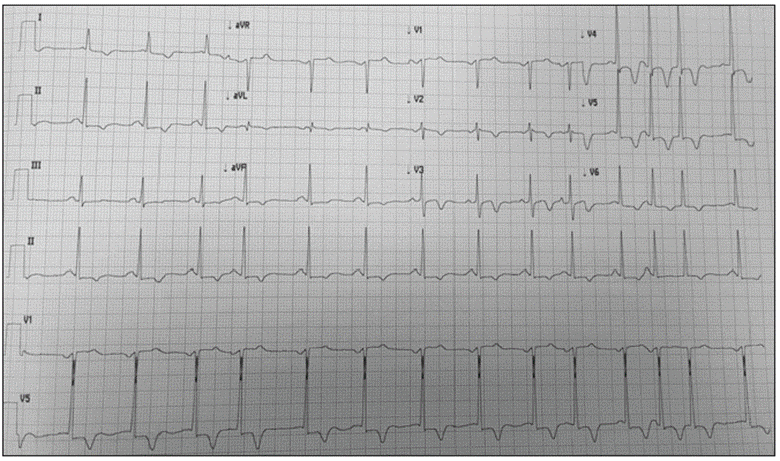
Figura 2 Signos de hipertrofia ventricular izquierda, con alteración de la repolarización y T negativa en derivaciones precordiales. Extrasistolia supraventricular y dupleta.
Por palpitaciones se realizó estudio de Holter de 24 horas con evidencia de complejos prematuros auriculares frecuentes y taquicardia auricular no sostenida.
Tras los hallazgos en el estudio ecocardiográfico en reposo se observó hipertrofia apical de ventrículo izquierdo (VI) que orientaba a los hallazgos de la electrocardiografía (miocardiopatía hipertrófica apical) basal. El Estudio no invasivo de isquemia coronaria (ecocardiograma de estrés ejercicio) fue negativo para la inducción de la misma durante el ejercicio y no tenía gradientes aumentados en el tracto de salida del VI.
Estudio de RM de corazón (Figura 3), confirmó hipertrofia asimétrica del ápex, espesor parietal apical durante la diástole de 17 mm y relación del espesor parietal/pared posterior de 1.54, con dinámica segmentaria y función sistólica global conservada. No se detectaron gradientes obstructivos a nivel del ventrículo izquierdo.
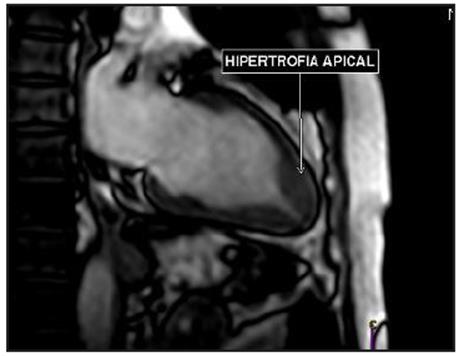
Se realizaron estudios para miocardiopatía hipertrófica descartando hipertensión arterial y tamizaje negativo para enfermedad de Fabry. Se planteó tratamiento sintomático a sus palpitaciones y extrasistolia auricular frecuente con betabloqueadores.
Debido a la ausencia de antecedentes familiares de muerte súbita cardiaca y bajo riesgo de muerte súbita calculado por escala de SCD HM de 2.5%, no se consideró candidato a implante de desfibrilador automático implantable. Paciente actualmente asintomático, con estudio negativo familiar en primer grado para MCH.
Discusión
El presente caso muestra el carácter silente de ciertas patologías que pueden implicar importante morbimortalidad si no se indica una intervención temprana. La miocardiopatía hipertrófica apical como variante de la mutación sarcomérica, conocida como síndrome de Yamaguchi, es una variante morfológica rara, en la cual no existe obstrucción al tracto de salida del ventrículo izquierdo sino obstrucción medioventricular 5. En el presente caso, el hallazgo de la patología fue incidental y diagnosticado a los 59 años. La mayoría de los pacientes se presentan con síntomas en su cuarta década de la vida, con una media de edad de presentación que oscila entre 41.4 ± 14.5 años siendo más frecuente en el género masculino 2. Nuestro paciente se diagnosticó a una edad más avanzada en comparación a lo reportado en la literatura, debido a que era asintomático y aunque en ocasiones manifestaba palpitaciones, eran leves y nunca fueron estudiadas.
En general, más de la mitad de los pacientes son asintomáticos como en el caso del diagnóstico de nuestro paciente, pero aquellos que presentan síntomas, pueden debutar con dolor torácico, palpitaciones, síntomas de insuficiencia cardiaca, disnea, fibrilación auricular o síncope 5.
La sospecha diagnóstica se inicia con la toma de un electrocardiograma basal. Las características electrocardiográficas típicas son la hipertrofia ventricular, la presencia de ondas T negativas gigantes (≥ 10 mm), depresión del ST y ondas U negativas en DII, III y aVF, V4-V6 y QTc prolongado. En el presente caso, estos hallazgos se encuentran compatibles con el diagnóstico. Los hallazgos más frecuentes son la clásica inversión profunda de la onda T en las derivaciones precordiales hasta en 93% de pacientes y cerca del 65% de ellos presentan hipertrofia ventricular izquierda. Las ondas T negativas gigantes, se han reportado en 47% de los pacientes con esta patología 5. No obstante, se ha descrito que las ondas T gigantes no se evidencian constantemente en esta entidad, dado que estas y la depresión del segmento ST pueden variar con el paso del tiempo 6.
El diagnóstico depende de la detección de hipertrofia apical localizada por medio de imagen y la ecocardiografía ha sido la primera línea para esto. Sin embargo, hay limitaciones para la correcta visualización del ápex y su hipertrofia, por lo que puede pasar inadvertida. Estas limitaciones son superadas a través de la RM de corazón, incluso en casos donde fueron omitidos por ecocardiografía 7. Los criterios diagnósticos por resonancia son: espesor de la pared apical mayor a 15 mm y/o una relación entre el espesor de la pared apical y basal del ventrículo izquierdo ≥ 1.3-1.5 8.
La miocardiopatía hipertrófica constituye un diagnóstico de exclusión, por lo cual, causas secundarias de hipertrofia ventricular izquierda como hipertensión arterial, estenosis aórtica valvular o subvalvular y miocardiopatía infiltrativa deben ser descartadas. Se realizó tamizaje para enfermedad de Fabry, que se ha estimado tiene una prevalencia de 0.51% en miocardiopatía hipertrófica y aunque la hipertrofia simétrica y concéntrica del ventrículo izquierdo es el patrón más típico en esta enfermedad, otras variantes como la apical, también se han reportado 9. Los patrones de otras enfermedades como amiloidosis o hemocromatosis por imagen y RM no fueron sugestivos, motivo por el cual no fueron investigados.
En cuanto al tratamiento, en la miocardiopatía hipertrófica, independientemente de los síntomas, la optimización del estilo de vida está recomendado, especialmente en cuanto a la educación sobre actividades aeróbicas de baja intensidad 10.
Se recomienda evitar el exceso de alcohol, sustancias psicoactivas, la deshidratación y temperaturas extremas como precipitantes de síncope o arritmias. El manejo inicial consiste en el uso de betabloqueadores para disminuir la frecuencia cardiaca y reducir el gradiente obstructivo en el tracto de salida del ventrículo izquierdo. La miectomía o la ablación septal con alcohol han sido métodos para aliviar la obstrucción al tracto de salida del ventrículo izquierdo, pero con evidencia limitada en la variante apical 5.
La muerte súbita cardiaca es menos probable que ocurra en pacientes con miocardiopatía hipertrófica apical aislada y la morbilidad cardiovascular general es menor comparada con otros fenotipos de miocardiopatía hipertrófica. Sin embargo, cerca del 25% de individuos pueden desarrollar eventos mórbidos significativos tardíos, incluyendo falla cardiaca, dolor torácico, fibrosis apical, formación de aneurisma apical, accidente cerebrovascular, fibrilación auricular y taquicardia ventricular 11. En Japón, la MCH apical parece ser más benigna comparada con los países occidentales, que suelen tener complicaciones como fibrilación auricular e infarto agudo de miocardio durante el seguimiento a largo plazo 7.
La guía de práctica clínica y los ensayos clínicos soportan el uso de desfibrilador cardioversor implantable (CDI) para la prevención primaria de muerte súbita (MS) en los siguientes casos: 1) historia familiar de muerte cardiaca súbita, 2) síncope no explicado, 3) taquicardia ventricular no sostenida asintomática, 4) respuesta anormal de la presión arterial al ejercicio, 5) espesor parietal del ventrículo izquierdo mayor a 30 mm. Sin embargo, se han identificado otros factores de riesgo potenciales como la obstrucción al tracto de salida del ventrículo izquierdo, el realce tardío con gadolinio extenso (fibrosis miocárdica), aneurisma apical, mutaciones genéticas/múltiples, espesor parietal del ventrículo izquierdo entre 25-30 mm 12.
La estratificación del riesgo se puede realizar por medio de la calculadora HCM risk-SCD 13 que incorpora la edad, extensión de la hipertrofia ventricular izquierda, tamaño de la aurícula izquierda, gradiente del tracto de salida del ventrículo izquierdo, historia familiar de muerte cardíaca súbita, taquicardia ventricular no sostenida y síncope inexplicado para predecir el riesgo a cinco años de muerte cardiaca súbita 1. Esta calculadora permite establecer en pacientes sin banderas rojas la necesidad del implante de un desfibrilador como prevención primaria. Es recomendado entonces de acuerdo al puntaje de riesgo la toma de decisiones así 12:
Puntaje mayor 6% se debe considerar implantar un desfibrilador (NE a COR).
Puntaje entre 4-6% (puede ser considerado).
Puntaje menor 4% no está generalmente indicado.
Finalmente tras una evaluación nuestro paciente obtuvo un puntaje de 2.5% no requiriendo hasta el momento del implante de un CDI. Cabe mencionar que actualmente no hay ensayos o modelos predictores que guíen este aspecto específicamente en MCH apical, aunque la escala anterior está basada en MCH con diferentes subtipos morfológicos, se ha considerado que en esta entidad específica puede subestimar el riesgo, por lo que se deben considerarse otros factores 14.
El tamizaje familiar fue negativo, lo cual concuerda con lo descrito en la literatura más actualizada en donde pocos pacientes reportan una historia familiar positiva comparado con la MCH clásica, lo que sugiere diferencias potenciales en el tamizaje y la presencia de otros factores etiológicos 14.
Aunque en Colombia el acceso al estudio de tamizaje tanto familiar como etiológico para miocardiopatía hipertrófica suele ser limitado, en nuestro caso el proceso por parte de la entidad de salud fue oportuno. Sin embargo, esto enmarca una oportunidad para mejorar las políticas de salud pública de nuestro país en este campo, que favorezcan el diagnóstico, tratamiento oportuno y que además involucren un estudio relacionado con esta entidad que permita conocer más su comportamiento a nivel local, pues incluso en la literatura actual, se le considera como la variante menos conocida 14.
Conclusiones
El síndrome de Yamaguchi, integra una variante rara de miocardiopatía hipertrófica en países occidentales. Constituye un reto diagnóstico, etiológico, pronóstico y terapéutico comparado con las otras variantes de MCH que son mejor conocidas, estudiadas y entendidas. Por este motivo, se requiere más investigación en este campo, sobre todo en países occidentales en donde sus complicaciones suelen ser mayores. La prevención primaria de muerte súbita debe considerarse igual que en las otras miocardiopatías hipertróficas con la búsqueda de banderas rojas y cálculo de riesgo, así como el alivio sintomático de la obstrucción del VI.
References
1. Geske JB, Ommen SR, Gersh BJ. Hypertrophic Cardiomyopathy: Clinical Update. JACC Hear Fail. 2018;6(5):364-75. [ Links ]
2. Yusuf SW, Bathina JD, Banchs J, Mouhayar EN, Daher IN. Apical hypertrophic cardiomyopathy. World J Cardiol. 2011;3(7):256-9. [ Links ]
3. Abugroun A, Ahmed F, Vilchez D, Turaga L. Apical hypertrophic cardiomyopathy: A case report. Kardiol Pol. 2017;8(5):265-8. [ Links ]
4. Guzmán MM, Ruth N, Cruz C, Mendieta CP, Mogo CC, López M, et al. Cardiomiopatía hipertrófica apical: Diagnóstico con resonancia magnética cardiovascular. Rev Invest Med Sur Mex. 2008;15(4):297-301. [ Links ]
5. Mirabbasi SA, Khalighi K, Mukkamala S, Kodali A. A rare case of apical hypertrophic cardiomyopathy (AHCM). J Community Hosp Intern Med Perspect [Internet]. 2017;7(2):122-5. Available from: https://doi.org/10.1080/20009666.2017.1324238 [ Links ]
6. Madias JE. Electrocardiogram in apical hypertrophic cardiomyopathy with a speculation as to the mechanism of its features. Netherlands Hear J. 2013;21(6):268-71. [ Links ]
7. Moon JCC, Fisher NG, McKenna WJ, Pennell DJ. Detection of apical hypertrophic cardiomyopathy by cardiovascular magnetic resonance in patients with non-diagnostic echocardiography. Heart. 2004;90(6):645-9. [ Links ]
8. Noureldin RA, Liu S, Nacif MS, Judge DP, Halushka MK, Abraham TP, et al. The diagnosis of hypertrophic cardiomyopathy by cardiovascular magnetic resonance. J Cardiovasc Magn Reson [Internet]. 2012;14(1):17. Available from: http://www.jcmr-online.com/content/14/1/17 [ Links ]
9. Caetano F, Botelho A, Mota P, Silva J, Marques AL. Fabiy disease presenting as apical left ventricular hypertrophy in a patient carrying the missense mutation R118C. Rev Port Cardiol [Internet]. 2014;33(3):183.e1-183.e5. Available from: http://dx.doi.org/10.1016/j.repc.2013.11.005 [ Links ]
10. Ommen SR, Mital S, Burke MA, Day SM, Deswal A, Elliott P, et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy: Executive Summary. Circulation. 2020;533-57. [ Links ]
11. Stainback R. Apical hypertrophic cardiomyopathy. Tex Hear Inst J. 2012;39(5):747-9. [ Links ]
12. Weissler-Snir A, Adler A, Williams L, Gruner C, Rakowski H. Prevention of sudden death in hypertrophic cardiomyopathy: Bridging the gaps in knowledge. Eur Heart J. 2017;38(22):1728-37. [ Links ]
13. Choi YJ, Kim HK, Lee SC, Park JB, Moon I, Park J, et al. Validation of the hypertrophic cardiomyopathy risk-sudden cardiac death calculator in the Asians. Heart. 2019;1-6. [ Links ]
14. Hughes RK, Knott KD, Malcolmson J, Augusto JB, Mohiddin SA, Kellman P, et al. Apical Hypertrophic Cardiomyopathy: The Variant Less Known. J Am Heart Assoc. 2020;9(5):e015294. [ Links ]
Recibido: 06 de Enero de 2021; Aprobado: 06 de Abril de 2021
 This is an open-access article distributed under the terms of the Creative Commons Attribution License
This is an open-access article distributed under the terms of the Creative Commons Attribution License
