











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkLa cristalografía de rayos X de monocristal es una técnica capaz de determinar con precisión la disposición atómica en el espacio tridimensional que conforman los materiales cristalinos [1]. Por medio de la interacción radiación-materia es posible la determinación de los parámetros posicionales y térmicos de los átomos que conforman una molécula en un sistema cristalino dado, lo cual permite elucidar completamente la estructura interna de los cristales [2].
Todos los cristales pueden describirse como una red tridimensional de puntos, con un grupo de átomos, moléculas o iones posicionados en cada uno de los puntos de la red. La porción más pequeña de la red se denomina celda unidad, la cual se repite ordenadamente a lo largo de las tres direcciones espaciales [2]; cuando esta se repite ilimitadamente en el espacio se forma la estructura cristalina [3]. Los parámetros de la celda unitaria se describen por la longitud de sus aristas (a, b y c) y los ángulos (a, p y y) formados entre ellas [3]. Existen muchas variedades de cristales, tanto naturales como sintéticos (diamantes, sal común, cuarzo, azúcar, etc.), y debido a sus diferentes propiedades físico-químicas, tienen innumerables usos en tecnología [4], farmacéutica [5], bioquímica, medicina [6] y procesos supramoleculares [7]. La formación de los cristales se debe a una combinación de interacciones covalentes y no covalentes como los enlaces de hidrógeno, ion-ion, ion-dipolo, dipolo-dipolo, entre otros [8]. Estas interacciones y la organización de la estructura cristalina son determinantes para entender las propiedades físicas y químicas de los diferentes materiales.
El análisis supramolecular de las superficies de Hirshfeld se ha convertido en una herramienta valiosa para analizar las interacciones intermoleculares; proporcionando información sobre los diferentes tipos de interacciones [9] no covalentes (ion-ion, ion-dipolo, dipolo-dipolo, de Van der Waals, interacciones ϖ-ϖ de apilamiento y puentes de hidrógeno) [10] presentes dentro de una estructura cristalina, ayudando a visualizar, explorar, analizar y cuantificar las interacciones para entender como las especies moleculares se mantienen unidas [8]. La formación de este tipo de superficies se basa en la partición del espacio cristalino en regiones, usando para eso el cociente entre las distribuciones electrónicas esféricas de cada átomo que la conforman (promolécula) y la suma de las densidades electrónicas de las moléculas vecinas que conforman el cristal (procristal) [11]. En la definición dada por Spackman y Byrom [11] se define la superficie de Hirshfeld con relación a la función de peso molecular,
la cual está dada por w(r ) =
 , indicando que la superficie es formada cuando la contribución de la promolécula a la densidad de electrones del procristal excede la de todas las demás moléculas del cristal w (r) ≥ 0,5. Lo cual garantiza un máximo de proximidad entre moléculas vecinas [12]. A partir de esta superficie se pueden determinar diferentes funciones de distancia desde la superficie de Hirshfeld de una molécula hasta el interior (di) o el exterior (de) de la superficie del átomo más cercano. Al graficar estos valores para una molécula dentro de un cristal, es posible obtener la huella digital (fingerprints plot) de las interacciones intermoleculares [13].
, indicando que la superficie es formada cuando la contribución de la promolécula a la densidad de electrones del procristal excede la de todas las demás moléculas del cristal w (r) ≥ 0,5. Lo cual garantiza un máximo de proximidad entre moléculas vecinas [12]. A partir de esta superficie se pueden determinar diferentes funciones de distancia desde la superficie de Hirshfeld de una molécula hasta el interior (di) o el exterior (de) de la superficie del átomo más cercano. Al graficar estos valores para una molécula dentro de un cristal, es posible obtener la huella digital (fingerprints plot) de las interacciones intermoleculares [13].
Un ejemplo de la aplicación de esta herramienta se plantea para el compuesto E-4-nitro- N’-(piridina-2-ilmetilen) benzohidrazida reportado por Gordillo et al. [14], el cual cristaliza en el sistema cristalino triclínico con grupo espacial P-1. Dentro de su unidad asimétrica se encuentran dos moléculas con características de confórmeros, presentando diferentes ángulos de torsión para los grupos nitro y amida. Además, esta estructura presenta interacciones de apilamiento tipo n-n entre los anillos piridínicos y de benceno. A partir de los gráficos de huellas dactilares fue posible encontrar las interacciones que más contribuyen a la superficie O…H (33,1%), H…H (29,7%) y C…C (12,1%) y las interacciones más fuertes se dan por contactos O…H y N…H.
El compuesto ácido 2- E -((4-hidroxifenil) diazenil) benzoico (Figura 1) es uno de los derivados del azobenceno utilizado como biomarcador, ya que cuando el grupo azo está conjugado con dos anillos aromáticos, el compuesto absorbe radiación electromagnética en el espectro visible presentando coloración. Es por esto que este tipo de compuestos son empleados como colorantes en la industria textil, papelera, alimentaria, etc. [15]. Debido a que estos compuestos tienen la capacidad de sufrir tautomerismo, se han realizado estudios de esta propiedad en el grupo azo-hidrazona en diferentes solventes con el fin de examinar la afinidad de unión con la estreptavidina combinando métodos espectroscópicos y computacionales [16]. Este compuesto puede ser utilizado para determinar la cantidad y la actividad de unión a la biotina de la avidina en solución, pues la avidina es una glucoproteína antagonista de la biotina, la cual se une con gran afinidad para formar un derivado insoluble que impide su biodisponibilidad [17]. Los derivados de azobenceno han sido investigados para su uso en el almacenamiento de datos digitales [18] como interruptores de encendido y apagado de dispositivos nanomoleculares y en canales iónicos [19].

Este trabajo está enfocado en el análisis estructural, supramolecular y de las superficies de Hirshfeld a partir de los datos de difracción de rayos X del compuesto ácido 2-(E)-((4-hidroxifenil) diazenil) benzoico. Para esto se resolvió la estructura con el programa SHELXT [20] mediante el método de fase intrínseca y se refinó por el método de mínimos cuadrados utilizando el programa SHELXL [21]. Posteriormente se realizó el análisis estructural para demostrar las diferencias estructurales entre los confórmeros observados en la unidad asimétrica, seguido de un análisis supramolecular mediante la utilización de superficies de Hirshfeld con el fin de analizar y cuantificar los diferentes tipos de contactos, además de obtener las redes energéticas que dan lugar al empaquetamiento cristalino. Con este trabajo, más que mostrar los resultados de investigación en cristalografía de pequeñas moléculas, se pretende mostrar de manera clara el proceso sistemático para la resolución, refinamiento y análisis de datos a partir de medidas de difracción de rayos X de monocristal, presentando una guía práctica para el análisis estructural y supramolecular.
Materiales y métodos
Los datos para la determinación de la estructura cristalina fueron obtenidos por difracción de rayos X de monocristal utilizando un difractómetro Rigaku XtaLab Mini con radiación Mo Ka (0.71073 Å) monocromada por grafito a temperatura ambiente (293 K). La determinación de la celda y los parámetros de celda finales se obtuvieron con todas las reflexiones usando el software CrysAlisPro [22]. La integración y el escalado de los datos se llevaron a cabo usando el mismo software.
Resolución y refinamiento de la estructura
La resolución de la estructura se realizó a partir de los datos de difracción, usando el software SHELXT [20] dentro de la plataforma de Olex2 [23]. Este método permitió determinar exactamente el grupo espacial y el mapa de Fourier, mostrando la ubicación de todos los átomos no hidrógenos. Posteriormente se realizó el refinamiento del modelo primario obtenido usando el método de mínimos cuadrados no lineales de matriz completa implementados en el programa SHELXL [21]. La estructura fue refinada, primero, isotrópicamente y, luego, anisotrópicamente, teniendo en cuenta los parámetros térmicos de cada átomo dentro del cristal. Finalmente se agregaron estereoquímicamente los átomos de hidrógeno que fueron refinados utilizando un modelo de átomo rígido (Uiso(H) = 1,2 eq para C-H aromáticos y 1,5 eq para O-H, con distancias fijas de 0,930 y 0,820 Å, respectivamente). Usando el software Mercury [24] se realizó el análisis estructural comparativo de las cuatro moléculas presentes en la celdilla unidad. Finalmente, con el Software Platón [25] se realizó la validación de los datos. Los datos cristalográficos están condensados en el archivo con formato .CIF (Crystallographic Information File) y se pueden encontrar en la base de datos CCDC bajo el código 1986080.
Análisis supramolecular: Superficies de Hirshfeld
Utilizando software Crystal Explorer [26] fueron generadas las superficies de Hirshfeld y los diagramas de huella dactilar (Fingerprint Plots) para el compuesto ácido 2-E-((4-hidroxifenil) diazenil) benzoico. El cálculo de las energías de interacción entre una molécula central de la unidad asimétrica con cada una de las moléculas vecinas fue realizado utilizando el programa TONTO [27] con un nivel de teoría DFT/B3LYP/6-31G(d,p). A partir de estos datos fue posible obtener los valores de energía de interacción total (Etot) y por componente, además de las redes energéticas del compuesto.
Resultados y discusión
Resolución estructural
La resolución estructural, entendida como la obtención de las fases asociadas a los factores de estructura a partir de las intensidades de difracción para la determinación de la densidad electrónica de cada átomo en el espacio, se realizó con el programa SHELXT [20] por el método de fase intrínseca. Este método utiliza una mezcla de métodos directos, superposición de Patterson y de espacio dual [28,29] a partir del formato estándar de SHELX (archivos .ins y .hkl). En el proceso de resolución, esta metodología construye de manera iterativa la estructura y puede asignar átomos erróneos que no tienen sentido químico, además de localizar algunas moléculas huésped con átomos también asignados incorrectamente [30] (ver Figura 2). SHELXT agrega datos no medidos por encima y por debajo del límite de resolución de los datos en el archivo HKL. Reduce los efectos de los errores de terminación en serie en las síntesis de Fourier, pero tiende a hacer que la integración de densidad electrónica utilizada para asignar los tipos de elementos sea menos confiable [20]. La fase intrínseca primero expande el conjunto de datos al grupo espacial P1 triclínico, usando los métodos directos, pero el proceso de la solución de la estructura inicia con una función de superposición de Patterson. Las fases se usan para determinar el grupo espacial con el propósito de mejorar el mapa de densidad electrónica inicial, luego realiza un refinamiento mediante el reciclaje del espacio dual con mapas de omisión aleatorios [20, 30]. La fase intrínseca asigna tipos de átomos al mapa de densidad electrónica final en las posiciones de mayor densidad electrónica (Figura 2) [20, 30].

Refinamiento
Una vez que se han asignado los átomos se realiza un refinamiento isotrópico utilizando una solución de gradiente conjugado de las ecuaciones normales de mínimos cuadrados [21]. Las densidades electrónicas son obtenidas por la aplicación de la trasformada de Fourier con el software SHELXL [21], el cual las ubica en el espacio y las nombra de mayor a menor densidad electrónica. A partir de ello y conociendo la identidad de los átomos presentes en el compuesto, se realiza una correlación átomo-densidad teniendo en cuenta tres variables posicionales y una variable térmica. Este análisis se denomina refinamiento isotrópico. Luego de obtener una estructura inicial bien definida, se realiza el refinamiento anisotrópico en el que se refinan nueve variables: tres posicionales y seis térmicas, teniendo en cuenta la vibración y el estado térmico de los diferentes átomos, lo cual es útil para obtener la estructura exacta de la molécula.
El refinamiento anisotrópico representa un modelo químico más razonable del sistema con respecto a la distribución de la densidad electrónica y potencialmente puede reducir las estadísticas de refinamiento de los índices de correlación R1 y wR2 (por ejemplo R1 en Figura 3), siempre que se utilicen datos con resolución adecuada y un buen factor de discordancia interno (Rint) [30]. Para un modelo de estructura bien refinado, el valor de R se aproxima a un valor pequeño, que corresponde a los errores en los datos experimentales y en el modelo [31]. El patrón de difracción se genera por la interacción de los electrones en el cristal con los fotones de rayos X [32]; sin embargo, los átomos más ligeros como el átomo de hidrógeno son difíciles de localizar, esto debido a que su baja densidad electrónica (un electrón) tiene un efecto muy débil sobre el patrón de difracción, por tanto, para posicionarlos durante el refinamiento de la estructura, los átomos de hidrógeno se localizan de manera manual y estereoquímica y teniendo en cuenta las distancias y enlaces aceptados, para así mejorar el refinamiento y obtener una estructura con sentido químico.

Análisis estructural
En la Figura 4 se presenta el diagrama ORTEP para el compuesto ácido 2-E-((4-hidroxifenil) diazenil) benzoico, donde se omitieron los átomos de hidrógeno para una mayor claridad. En la Tabla 1 se encuentran los datos cristalográficos y de refinamiento para el mismo compuesto.

Figura 4 Diagrama tipo ORTEP con un 50 % de probabilidad del compuesto ácido 2-E-((4-hidroxifenil) diazenil) benzoico. Los átomos de hidrógeno fueron omitidos por claridad.
Tabla 1 Datos cristalográficos y de refinamiento para el compuesto ácido 2-E-((4-hidroxifenil) diazenil) benzoico.

El compuesto ácido 2-E-((4-hidroxifenil) diazenil) benzoico cristaliza en el sistema cristalino ortorrómbico con grupo espacial Pbca. Dentro de la unidad asimétrica se encuentran cuatro confórmeros denominados I, II, III y IV (Figura 5), ya que presentan diferencias estructurales entre ellos (Tabla 2). Se puede observar que los ángulos diedros formados entre los planos que pasan por los anillos aromáticos son diferentes para cada molécula, al igual que el ángulo de torsión entre el anillo aromático y el carbonilo (Tabla 2). También es posible observar interacciones intramoleculares de hidrógeno O…H…N con diferente distancia para cada uno de los confórmeros (Tabla 2).

Figura 5 Confórmeros observados en la unidad asimétrica para el compuesto ácido 2-E-((4-hidroxifenil) diazenil) benzoico. Ejemplos de ángulo diedro entre planos de los anillos aromáticos y ángulo de torsión entre el anillo y el carbonilo para el confórmero II.

Se realizó la validación de los datos obtenidos de la estructura resuelta usando el programa Platón [25], el cual informa aspectos acerca de la consistencia e integridad de los datos reportados en el archivo .CIF. Se observó que el refinamiento de la estructura presenta varias alertas tipo A, B y C (siendo las alertas tipo A las más importantes); la alerta tipo A se debe principalmente al elevado valor del Rint (0,407) que debería ser menor a 0,10. Esta alerta está relacionada directamente con la baja cristalinidad del cristal estudiado así como con su tamaño, dado que los mismos presentaban morfologías de agujas con dos dimensiones muy pequeñas (menores que 10 μm). La combinación de ambos efectos perjudicó fuertemente el poder de difracción de la muestra, presentando intensidades observadas (se denomina intensidades observadas a aquellas reflexiones cuyas intensidades son 3 veces mayores que su ancho a media altura, I>3σ(I)) más allá de una resolución de 25° en 2θ. Esto afectó claramente la calidad de los datos resultantes y, por ende, se vio afectada la calidad del refinamiento, aunque el modelo propuesto presente una muy buena concordancia química [32].
Análisis supramolecular
El análisis supramolecular del compuesto ácido 2-E-((4-hidroxifenil) diazenil) benzoico fue realizado con ayuda del software Mercury [24] con el cual fue posible observar las diferentes interacciones supramoleculares que dan lugar a la formación del empaquetamiento cristalino (Figura 6). En la celda unidad se encuentran treinta y dos moléculas que forman hélices β a lo largo del eje [010] (ver Figura 6 a). Las hélices β se forman por interacciones de hidrógeno en la dirección [100] entre los confórmeros I y III, por interacciones O9…H9…O1 (i) y O3-H3…O7 (ii) con distancias de 2,799 (10) Å y 2,769 (10) Å respectivamente y un ángulo de 159° para cada una. Entre los confórmeros II y IV (ver Figura 6 a), con interacciones O6-H6…O10 (ii) y O12-H12--O4 (i) con distancias de 2,784 (10) A y 2,793 (10) Å respectivamente y ángulos de 164° y 169°.

Figura 6 Interacciones a lo largo de la dirección a) [010], b) [001] y c) [100] para el compuesto ácido 2-E-((4-hidroxifeml) diazenil) benzoico.
En la dirección [100] se observan interacciones débiles de tipo C38-H38--O2 (iii) y C23-H23…O11 (ii) con distancias de 3,353 (12) A y 3,281 (13) Å, y ángulos de 141° y 163°, respectivamente (Figura 6 c). En la dirección [001] (Figura 6 b) se observan interacciones de hidrógeno no clásicas entre los confórmeros II y III, por interacciones C30-H30…O6 (iv) con una distancia de 3,422 (13) A y un ángulo de 150°; y entre los confórmeros I y II, por interacciones C16-H16…O1 (v), C3-H3a…O4 (v) y C17-H17…O9 (vi), con distancias de 3,405(3) Å, 3,439(5) A y 3,486(4) Å, y ángulos de 133,97°, 141,55° y 153,55°, respectivamente.
Estas interacciones se dan entre las hélices beta a lo largo del eje [001]. La combinación de todas las interacciones en las tres direcciones da lugar al empaquetamiento cristalino del compuesto. ((i) -1/2+x,y,1/2-z; (ii) -x,1/2+y,1/2-z; (iii) 1/2+x,y,1/2-z; (iv) x,1/2-y,1/2+z; (v) 1/2+x,1/2-y,1-z; (vi) x,1/2-y,-1/2+z).
Análisis supramolecular mediante superficies de Hirshfeld
Utilizando el software Crystal Explorer [26] fue posible calcular las superficies de Hirshfeld y los diagramas de huella dactilar con el fin de cuantificar las interacciones que dan lugar a la formación del empaquetamiento cristalino (Figura 7). Las superficies de Hirshfeld son representaciones de las distancias intermoleculares en términos de los radios de Van der Waals, en donde las distancias menores a la suma de los radios de Van der Waals se representan en rojo, los valores iguales a la suma se representan en blanco y las distancias mayores se representan en azul (Figura 7 a). Los valores de las interacciones se pueden cuantificar usando las huellas dactilares (Figura 7 b); en esta se pueden observar, para el Confórmero IV, picos más pronunciados y con valores bajos de de + di para los contactos de O…H/H…O (Figura 7 (c)), lo que indica la presencia de un tipo de contacto fuerte. En los contactos para la Figura 7 (d-e) los valores observados se encuentran en una región con valores más altos de de + di que corresponden a interacciones más débiles.

Figura 7 a) Superficie de Hirshfeld, b) Diagrama de huella dactilar general y c-e) de las interacciones más representativas para el Confórmero IV del compuesto ácido 2-E-((4-hidroxifenil) diazenil) benzoico.
Se pudieron observar los diferentes tipos de interacciones intramoleculares e intermoleculares de cada confórmero donde se encuentra bastante similitud entre ellos (Figura 8), predominando las interacciones tipo H…H con un 34,1% para los confórmeros I y III, y 32,2% para los confórmeros II y IV, seguidas de contactos tipo O…H/H…O y C…H con porcentajes menores en el valor total de la superficie.

Se realizó una búsqueda en la base de datos CCDC [33], donde se encontró una molécula (WAMFIA) estructuralmente similar al compuesto de estudio (AEHDB) [34]; en la Tabla 3 se reportan los datos cristalográficos para el compuesto denominado WAMFIA. En la Figura 9 se compararon los diagramas de huellas dactilares de ambos compuestos, donde se observa que los principales contactos están dados por interacciones tipo H…H con porcentajes de contribución total a la superficie supramolecular de 34,8% para AEHDB y 36,8% para WAMFIA. También son observados contactos tipo O…H/H…O y C…H (Figura 9) con contribuciones altas a la superficie total. Cabe resaltar que WAMFIA cristaliza en el grupo espacial P2 1 2 1 2 1 , con una molécula del derivado de azobenceno y una molécula de agua en su unidad asimétrica [34]; mientras que AEHDB cristaliza en el grupo espacial Pbca, con cuatro moléculas tipo confórmeros en su unidad asimétrica. Esta diferencia, aunque no es muy notoria en los porcentajes de los principales contactos en cada fase cristalina, se ve reflejada claramente en el perfil de los diagramas de huella dactilar para cada fase y en las interacciones C…C y N…H/H…N, las cuales presentan diferencias significativas, siendo mayores para el compuesto WAMFIA.


Figura 9 Porcentaje de interacciones calculadas para el compuesto ácido 2-E-((4-hidroxifenil) diazenil) benzoico │≠(AEHDB) y un sistema cristalino semejante (WAMFIA).
Las gráficas de huellas dactilares (fingerprint) bidimensionales para las interacciones más significativas de WAMFIA se muestran en la Figura 10, donde se observan las interacciones O…H/H…O (Figura 10) con picos muy definidos, evidenciando contactos de hidrógeno fuertes (de+di=1,7). Para las demás contribuciones a la superficie total no se observan picos definidos, presentando una diferencia sustancial entre los diagramas de huella dactilar de ambos compuestos.

Análisis de interacciones energéticas
Las energías de interacción (Etot) son calculadas con precisión, entre una molécula central de la unidad asimétrica con cada una de las moléculas vecinas, para obtener la interpretación supramolecular de las interacciones presentes, las cuales llevan a la formación del cristal y sus redes energéticas [9]. Estas energías (Etot) se expresan en términos de energía electrostática (Eele), polarización (Epol), dispersión (Edis) y repulsión (Erep), según el modelo propuesto (Etot= Eele+Epol+Edis+Erep) [35]. Las energías de interacción para el compuesto ácido 2-E-((4-hidroxifenil) diazenil) benzoico fueron calculadas a un nivel de teoría DFT/B3LYP/6-31G(d,p). Los valores obtenidos se presentan en la Tabla 4, donde N es el número de moléculas que comparten la misma simetría y energías de interacción equivalentes y R es la distancia entre los centroides moleculares (posición atómica media) en A. Las energías para un grupo de moléculas alrededor de una molécula seleccionada se calculan en un radio de 3,8 A, apropiado para la mayoría de los cristales orgánicos [36].
Tabla 4 Energías de interacción (kJ/mol) calculadas para la formación del cristal por B3LYP/6-31G(d,p).

*Los factores de escala de las densidades electrónicas para el modelo usado (B3LYP/6-31G(d,p)) son: Eele: 1,057, Epol: 0,740, E4s: 0,871 y Erep: 0,618. Se usa un color para ayudar en la identificación de la energía de interacción de cada molécula en particular con la molécula central (Tabla 4 y Figura 11).
La energía total más alta de interacción (Etot) es -33,3 kJ/mol entre la molécula central de la unidad asimétrica y la molécula naranja al costado (Figura 11), donde la mayor contribución se da en términos de energía de dispersión (Edis = -41,6 kJ/mol), seguida de una menor energía electrostática (Eele = -6,9 kJ/mol). La primera interacción energética se debe a contactos débiles de Van der Waals y la segunda a posibles contacto dipolo-dipolo. La segunda interacción más fuerte tiene una energía Etot = -28,1 kJ/mol, la cual tiene la componente de energía de dispersión más alta de todas las energías calculadas Edis= -57,5 kJ/mol; lo cual es consecuencia de los contactos tipo H…H, tal como se observó anteriormente en las huellas dactilares donde las contribuciones predominantes provenían de este tipo de contactos. Cabe anotar que, en ambos casos, los valores de energía de repulsión están entre 18,2 a 22,5 kJ/mol, los cuales afectan la estabilidad del empaquetamiento cristalino.
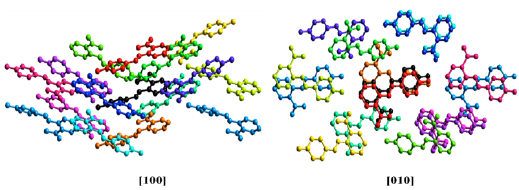
Figura 11 Empaquetamiento cristalino alrededor de una molécula central (Negro) vista desde las direcciones [100] y [010]. El color en cada molécula está relacionado con los valores de energía de la tabla 4 (los átomos de hidrógeno fueron omitidos por claridad).
La red de energías vecinas más cercanas muestra las interacciones intermoleculares en un cristal, mediante un marco de cilindros cuyo ancho es proporcional a la fuerza de la interacción [37]. Los cilindros que componen el marco representan las fortalezas en el empaquetamiento molecular. Es por esto que, para el compuesto ácido 2- E-((4-hidroxifenil) diazenil) benzoico, se observa que las energías que más contribuyen a la energía total son la energía de dispersión y energía electrostática (Figura 12), debidas a las interacciones H…H e interacciones de hidrógeno respectivamente. Estos valores están de acuerdo con lo observado a partir de las huellas dactilares, donde la principal contribución a la superficie general se da por los contactos débiles de H…H, seguido de interacciones O…H/H…O.

Figura 12 Marcos energéticos de las interacciones que más contribuyen a la energía de interacción total Etot para el compuesto ácido 2-E-((4-hidroxifeml) diazenil) benzoico (tamaño del tubo 100 unidades, Se toma como cut-off los valores de energía reportados en la figura. Los átomos de hidrógeno fueron omitidos por claridad).
Conclusiones
El compuesto ácido 2-E-((4-hidroxifenil) diazenil) benzoico cristaliza en el sistema ortorrómbico con parámetros de celda de 22,021(9) Å, 16,567(6) Å, 25,555(14) Å, y grupo espacial Pbca. En la unidad asimétrica se encontraron cuatro moléculas con características de confórmeros, ya que se pudieron evidenciar sus diferencias estructurales como los ángulos diedros entre los anillos, el ángulo de torsión en el grupo ácido carboxílico y enlaces de hidrógeno intramoleculares, que son diferentes para cada una de ellas. La validación de los datos en el software Platón mostró una alerta principal tipo A debido al alto valor obtenido del Rint (0,407); los datos usados para el análisis fueron obtenidos de un cristal muy pequeño y de baja calidad, lo cual genera valores estadísticos altos cuando se consideran todas las reflexiones (wR2), aunque el modelo reportado presente buena concordancia química y un aceptable índice de correlación R1. Se observó que en cada uno de los confórmeros se favorecen los enlaces intramoleculares O…H…N entre el ácido carboxílico y el grupo azo, lo cual estabiliza la configuración E del compuesto. Otra diferencia observada entre los confórmeros está en las gráficas de huellas dactilares bidimensionales, observando diferentes porcentajes de contribución en todos sus tipos de contactos. Desde el punto de vista del empaquetamiento cristalino y a lo largo de la dirección [010], se observó la formación de hélices beta por medio de interacciones de hidrógeno que se dan en la dirección [100], entre los confórmeros I y III y entre los confórmeros II y IV. Las cadenas de las hélices presentan interacciones de puente de hidrógeno clásicas y no clásicas, entre ellas en la dirección [001] y [100]; lo que da lugar a la formación del empaquetamiento cristalino. Con las superficies de Hirshfeld y las redes de interacciones energéticas tridimensionales, se determinó que las interacciones responsables del empaquetamiento cristalino son principalmente interacciones de dispersión, seguidas de enlaces de hidrógeno. Este comportamiento indica que, aunque las interacciones de dispersión son débiles debido a la gran cantidad de interacciones presentes en la molécula, estas contribuyen a la estabilización y formación del empaquetamiento cristalino. Finalmente, con este trabajo se consigue mostrar el tratamiento sistemático en cuanto a tratamiento de datos que involucran resolución estructural, refinamiento, validación y análisis supramolecular para un compuesto orgánico.

