











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
Desde hace 20.000-30.000 años A. de C., el mercurio ha sido un elemento incluido en la mano de obra del hombre [1], siendo la minería la más reconocida (exactamente en la extracción de oro a base de mercurio) y, a la vez, la actividad que más contaminación provoca por este metal. Por tanto, durante todo este tiempo ha surgido bioacumulación en su mayor reservorio (ríos, lagos y océanos), provocando afectaciones ambientales y en el ser humano [2]. La bioacumulación en los océanos es debida, por lo general, a las bacterias acuáticas anaeróbicas reductoras de sulfato, que transforman el mercurio elemental a metilmercurio por medio de la biometilación de este elemento [3], siendo el estado más contaminante del mercurio; aunque también puede ser transformado por actividades antropogénicas y ser, posteriormente, llevadas a las zonas acuáticas naturales. Afectando de manera directa a organismos acuáticos, y luego a las personas quienes basan su dieta principalmente de productos marinos [4].
En sintonía con la evolución del bienestar ambiental y humano, han surgido diferentes soluciones para este efecto adverso, como lo es la biorremediación, un mecanismo liderado por microorganismos y sus enzimas, para la degradación y transformación de contaminantes a otro estado de oxidación, el cual es menos tóxico para el medioambiente [5]. Entre estos microorganismos se encuentran las bacterias, uno de cuyos géneros es el Pseudomona [6]; las bacterias de este género son organismos Gram negativos de vida libre, con genomas grandes (en promedio 6 Mb), en los cuales tienen la información para adaptarse fácilmente a las condiciones que el ambiente en el que se encuentra le presente. Una de las especies de este género, con una amplia capacidad de adaptación y que ha sido usada comúnmente en procesos de biorremediación, es el Pseudomonas fluorescens. Además, se ha reportado que este microorganismo ha logrado sobrevivir en lugares con alta concentración de mercurio, por lo que la identificación de los elementos genéticos presentes, y que le dan esta característica, se han convertido en un importante tema de estudio [7].
Se ha identificado que los sistemas de resistencia bacteriana a mercurio se agrupan en un operón, conocido como el "operón Mer", que varía en estructura y consta de genes que codifican proteínas para la regulación (merR, merD), transporte (merT, merP and/or merC, merF) y reducción de mercurio (MerA) [8] y, en algunos casos, un gen adicional, merB, el cual otorga a los microorganismos que lo presentan la capacidad de cortar el enlace carbono-mercurio de organomercuriales como el acetato fenilmercúrico, y generar Hg2+, que luego es desintoxicado como mercurio metálico por la reductasa mercúrica. Se ha identificado la presencia de este gen en la bacteria P. fluorescens, como uno de los elementos importantes de su capacidad de biorremediación de mercurio [9-11]. MerB es la encargada de escindir el enlace carbono-mercurio por medio de una protonólisis produciendo un compuesto alifático o aromático y Hg (II) [12], el cual posteriormente es reducido a Hg (0) por la reductasa específica MerA [13]. MerB muestra una amplia especificidad de sustrato y puede dividir una gran variedad de compuestos organomercuriales que van desde compuestos de alquilo simples como Me-Hg a poliaromáticos y compuestos heterocíclicos como la merbromina [14]. En diversos reportes se evidencia que, para llevar a cabo esta reacción catalítica, los sitios activos de la proteína MerB varían en los diferentes macroorganismos en los que esta ha sido estudiada, como lo es en el dominio bacteria y en las arqueas. Se han identificado mutaciones génicas que en algunos casos producen cambios en la ubicación de algunos aminoácidos; aunque, en general, en la molécula se mantiene una gran similitud. Sin embargo, se ha identificado que estas variaciones ocasionan cambios en la expresión génica y en la estructura proteica [15].
Son estos últimos elementos los que sustentan la importancia de la biología estructural y la química computacional, con el fin de establecer con exactitud la estructura de las proteínas, confiriendo mayor conocimiento de su estructura, funcionalidad, su dinámica, las interacciones con ligandos y otras proteínas [16, 17]. Tal estudio se realiza por medio de herramientas proteómicas, con el propósito de observar las estructuras proteicas como lo es la cristalización con ayuda de la difracción de rayos X o de resonancia magnética, donde la calidad de la imagen estructural final es directamente proporcional a la perfección de las propiedades físicas del espécimen cristalino, que otorga con precisión las coordenadas atómicas, necesitándose cristales de gran tamaño y calidad suficiente para lograr obtener una recopilación de datos precisa. Sin embargo, este tipo de tecnologías son complejas y muy costosas, por lo que para todas las proteínas identificadas no existen datos de estructura generados experimentalmente [18, 19]. Pero más allá de ser un limitante, también impulsó el desarrollo de la química computacional, la cual, hoy en día, se orienta al modelaje y evaluación de proteínas in silico, una variante viable para ahorrar tiempo en el diseño de experimentos con mejores expectativas de éxito y disminuir costos en materiales y reactivos. Entre estas herramientas se encuentra el "modelado por homología", también conocido como "modelado comparativo" o "modelado basado en plantilla (TBM)"; estas estrategias de modelado de la estructura 3D de una proteína, usan una estructura conocida experimentalmente como molde.
En el presente artículo se plantea estudiar, con el uso de herramientas bioinformáticas, el gen, la secuencia de proteína y dar a conocer una estrategia para modelar y analizar la estructura tridimensional de la enzima MerB en Pseudomonas fluorescens, la cual no ha sido reportada. De igual manera, se plantea llevar a cabo la caracterización de la estructura secundaria, identificación de dominios funcionales, estructurales y la especificación del centro activo de esta importante molécula básica en los procesos de biorremediación de mercurio.
Materiales y métodos
Secuencias de MerB y alineamiento múltiple
Las secuencias de la familia de MerB en varias especies del género Pseudomonas, se obtuvieron de la plataforma de UniProt [20], estas se usaron para la realización de un alineamiento múltiple con la aplicación del European Bioinformatics Institute [21], T-Coffe [22], con el fin de identificar el nivel de diversidad entre ellas y, por tanto, la homología presente.
Modelamiento de la estructura tridimensional de MerB de Pseudomonas fluorescens
Con la secuencia identificada para MerB de Pseudomona fluorescens, se desarrolló un modelamiento por homología con la aplicación Geno3D [23], donde, en primer lugar, se introduce la secuencia y se corre con los valores exactos que propone la aplicación, con el fin de seleccionar el modelo con mejor valor propuesto, donde luego se extrae su respectiva secuencia y para facilitar su visualización se introduce en el software RasMol [24].
Análisis de la estructura secundaria de MerB de Pseudomonas fluorescens
Se predijo la estructura secundaria de MerB mediante la aplicación PSIPRED [25], la cual utiliza un método altamente preciso para la predicción de la estructura secundaria a partir de secuencias de aminoácidos.
Evaluación y validación de la estructura tridimensional de MerB de Pseudomonas fluorescens
Se evaluó la validación de los modelos predichos usando los programas Biovia [26] para verificar la validez de su estructura secundaria; ProSA-Web [27] para su ajuste a estructuras predichas mediante métodos de laboratorio, como la resonancia magnética nuclear o la difracción de rayos X; y Verify 3D [28], con el fin de llevar a cabo la validación de los modelos de los perfiles tridimensionales.
Identificación de dominios proteicos
Los dominios proteicos de la secuencia de MerB en P. fluorescens, se identificaron por medio de la página web de búsqueda MotifSearch [29].
Resultados
Alineamiento por pares y múltiple
A partir de las secuencias de la familia de las proteínas MerB (Tabla 1), se realizó, en la aplicación Phylogeny.fr [30], el alineamiento múltiple y la obtención del respectivo árbol filogenético, identificando en él las secuencias de MerB para P. fluorescens, y se obtuvieron cuatro secuencias en diferentes posiciones de la filogenia, como se muestra en la Figura 1.
Tabla 1 Código y organismos dei género Pseudomona spp aplicados en el estúdio filogenético, obtenidos de UniProt [20].
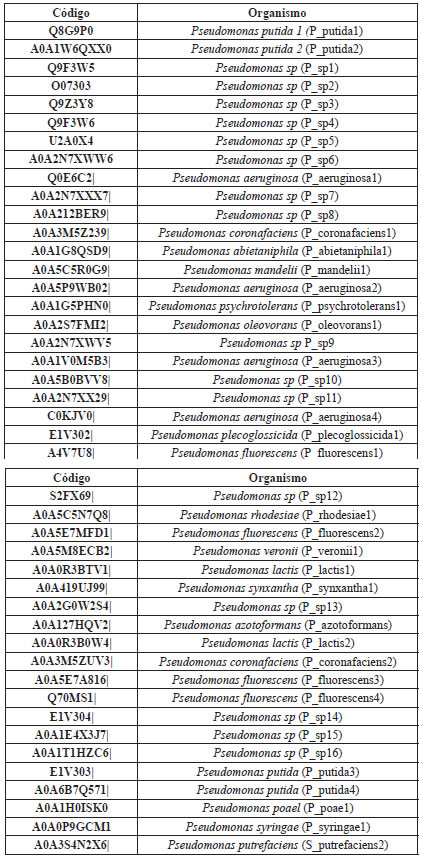

Figura 1 Árbol filogenético producto del alineamiento en Phylogeny.fr [30] de las secuencias reportadas de Pseudomonas para MerB (se resaltan en color azul los aislados de P. fluorescens, en rojo se observan los valores de Bootstrap, con 500 repeticiones, verificando la estabilidad de cada agrupamiento).
A partir del árbol filogenético se observan las diferentes secuencias de MerB de Pseudomona fluorescens, y se identifica la diversidad entre ellas, especialmente la secuencia "P fluorescens 2", que tiene mayor similitud con P syringae, y es diferente a los demás representantes de su especie en, al menos, el 5%. Teniendo en cuenta este nivel de diferencia con las otras secuencias correspondientes en la especie, se escogió la secuencia representativa para MerB de P. fluorescens para el análisis del modelamiento por homología.
Caracterización fisicoquímica de la enzima MerB de P. fluorescens
Partiendo de la secuencia proteica seleccionada de A0A5E7MFD1 para MerB identificada en P fluorescens (212aa), se verificó por medio de la plataforma Expasy [31] que su peso molecular es de 23139,4 Da y su punto isoeléctrico es de 5,86. Mediante la plataforma Uniprot [20] se estableció su código EC 4.99.1.2, correspondiente a una liasa de alquilmercurio, para la cual su reacción enzimática se presenta en la Figura 2.

Figura 2 Reacción enzimática de MerB identificada para P. fluorescens, dando lugar a una reacción catabólica teniendo como sustratos un alquilmercurio y un protón de hidrógeno, que produce un alcano y un ion mercurio. Tomada de Uniprot [20].
Modelamiento de la estructura tridimensional de la proteína MerB de P. fluorescens
Con los resultados suministrados por Geno3D, se estableció que la mejor secuencia para el modelamiento por homología es la proteína MerB en complejo con mercurio de Escherichia coli 3FN8 (Figura 3). Esta secuencia presentó mayor similitud a la secuencia MerB de P. fluorescens 2 con un valor E de 2 x 1050 y una desviación media de 0,48 Å.
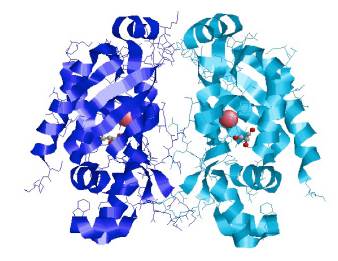
Figura 3 Modelo 3FN8 computacional generado a partir del estudio por homología para MerB en P. fluorescens a través de RasMol [24], observándose una proteína dimérica, cada cadena con un centro activo de mercurio (esferas rosa) y glicerol (estructura atómica), unidos covalentemente a residuos de aminoácidos.
Cada centro activo se logra (observar en la Figura 4) formado por un complejo de mercurio, el cual es clasificado como un complejo metálico, donde se sitúa un ion de mercurio (Hg+2) unido covalentemente mediante enlace metálico a dos cisteínas (Cys 96 y Cys 159) y a un ácido aspártico (Asp 99) y por medio de un puente de hidrógeno a un glicerol (Gol 222).

Figura 4 Ilustración computacional del complejo metálico de mercurio para MerB de E. coli 3FN8. Tomada de RCSB PDB Protein Data Bank [32].
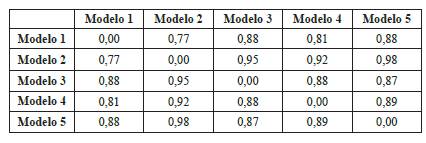
Tomando a 3FN8 como molde para el diseño de MerB de P fluorescens, se generaron cinco modelos (Tabla 2), donde los valores de correlación entre los cinco modelos evaluados se determinaron mediante desviación de la raíz cuadrada media (DRCM) con valores en ángstroms (Tabla 3).
Tabla 2 Comparación de la calidad de los modelos producidos por el servidor Geno3D. Todos los modelos utilizaron el mismo molde 3FN8. Cada columna representa: los valores de energía en Kcal/mol, el porcentaje de residuos que se encuentran en la región nuclear y el porcentaje de residuos no permitidos en el diagrama de Ramachandran. Los valores de energía indican la fiabilidad y la precisión de la estructura.

A partir de los valores obtenidos, se escogió el mejor modelo, el cual corresponde al valor negativo más bajo: el modelo 4; además, tiene porcentajes permitidos en el gráfico de Ramachandran.
El modelo se observó mediante la aplicación RasMol, dando como resultado la proteína presentada en la Figura 5.

Figura 5 Estructura proteica propuesta, modelo 4, para MerB de P. fluorescens. Se observa un monómero, con una estructura estable, que presenta hojas beta, hélices y pequeños giros. Obtenido de RasMol [24]. Al lado derecho se muestra el monómero correspondiente a la estructura usada como molde para el diseño propuesto.

Figura 6 Gráfico de secuencia y relación con la estructura secundaria para la enzima de MerB de P. fluorescens, utilizando la aplicación PSIPRED [25]. En amarillo, se identifican los residuos con tendencia a formar hojas beta; en rosa, los residuos con tendencia a formar hélices alfa; y en gris, se muestran los residuos sin tendencia específica a formar una estructura secundaria.

Figura 7 Diagrama de Ramachandran desarrollado por el programa Biovia [26] del modelo 4, en el que se muestra la presenciade cadauno de los aminoácidosde la secuencia en las regiones permitidas y no permitidas para su respectiva estructura secundaria.
Evaluación y validación de la estructura secundaria para MerB de Pseudomonas fluorescens
Para el análisis de la estructura secundaria, PSIPRED identifica nueve tramos de aminoácidos con tendencia a formar láminas beta y siete tramos con tendencia a hélices alfa (Figura 6).
Con la herramienta Biovia se verificó, por medio del gráfico de Ramachandran, la estructura propuesta en la Figura 6, identificando los aminoácidos ubicados en la región de valores permitidos y no permitidos en su estructura secundaria y la distribución de los giros Psi/Phi para el modelo 4, homóloga a P. fluorescens 2 (Figura 7). El diagrama de Ramachandran [33] dado para el modelo 4 de MerB, para P. fluorescens reveló que posee una estructura secundaria con una conformación estable, teniendo el 72,4% de residuos en las regiones más favorables, 21,6% como residuos permitidos, 4,3% en regiones generosamente permitidas y en las zonas no permitidas se encuentra una minoría de residuos del 1,6% diferentes a glicinas, para dar un total de 100% de residuos ubicados en el gráfico.
Los aminoácidos en el cuadrante superior izquierdo correspondiente a láminas betas paralelas, antiparalelas y giros, logran una mayor cantidad de residuos de hélices alfa con giro hacia la derecha en el cuadrante inferior izquierdo y un porcentaje mínimo de residuos en el cuadrante superior derecho conformados por hélices alfa con giro hacia la izquierda. Además, se identificaron que 185 residuos son diferentes a glicina-prolina, dos son los residuos terminales (exactamente glu y pro), el número de glicinas representado en triángulos es de 11, y el número de prolinas fue es de 11 para un total de 209 aminoácidos.
Entre su estructura se encuentran los motivos mostrados en la Figura 8, postulados por NCBI-CDD y Pfam, descritos en las Tablas 4 y 5, con un límite de valor E de 1,0.

Figura 8 Motivos obtenidos a partir de la plataforma MotifSearch [29] mediante las bases de datos NCBI-CDD y Pfam para la secuencia proteica MerB de P. fluorescens.
Tabla 4 Descripción de motivos resultantes del análisis en MotifSearch [29] mediante la base de datos Pfam para la secuencia proteica MerB de P. fluorescens.

*Primer número: aminoácido donde inicia su posición en la secuencia proteica de MerB.
(..): representativo de la contracción "al".
Segundo número: aminoácido donde termina su posición en la secuencia proteica de MerB.
Tabla 5 Descripción de motivos resultantes del análisis en MotifSearch [29] mediante la base de datos NCBI-CDD para la secuencia proteica MerB de P. fluorescens.

*Primer número: aminoácido donde inicia su posición en la secuencia proteica de MerB.
(..): representativo de la contracción "al".
Segundo número: aminoácido donde termina su posición en la secuencia proteica de MerB.
Entre los motivos encontrados, se escogieron aquellos con un valor E más bajo: el motivo MerB con mayor tamaño en la secuencia con ubicación en el aminoácido 79 al 1.999 con un valor E de 2,3 10-36, seguidamente de HTH_15 con ubicación 8 al 76 y un valor E de 8,4 10-23.
Validación de la estructura tridimensional propuesta para la proteína MerB de Pseudomonas fluorescens
Con el objetivo de validar el modelo 4 postulado como el homólogo de MerB para P. fluorescens, se desarrolló un análisis de calidad general del modelo, utilizando la plataforma Prosa-Web. Como resultado, esta estructura evaluada se calificó con un puntaje Z de -6,5, y se encuentra dentro del rango de puntuaciones típicas de las proteínas nativas de este grupo proteico (Figura 9). Se observa, de igual manera, que la estructura propuesta se encuentra dentro de la sección óptima en la figura del lado izquierdo, verificado por el punto negro. En la figura del lado derecho, se representa la calidad local del modelo por medio de la energía basada en conocimiento; para este análisis se identificó la presencia de picos que corresponden a regiones de paso a través de la membrana de la proteína, lográndose así una estructura totalmente sin partes problemáticas o erróneas en el modelo, evidenciándose en los valores negativos correspondientes.
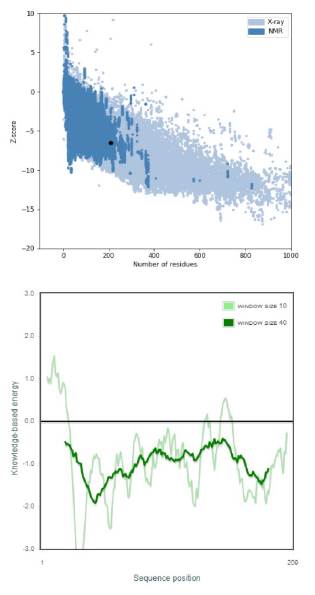
Figura 9 Datos obtenidos de validación de la estructura tridimensional del modelo 4 mediante la aplicación Prosa-Web [27]: a) Calidad total del modelo a través del puntaje Z (Z-score) y b) Calidad local del modelo por medio de la energía basada en conocimiento.
En seguida, se confirmaron los datos obtenidos de Prosa-Web con la herramienta Verify 3D (Figura 10), mediante un análisis donde el límite máximo esperado es de 100,00%, de los residuos tienen una puntuación 3D/1D promedio > = 0,2. Para el modelo propuesto (modelo 4), se identificó que este presenta el 80% de los aminoácidos que han puntuado con valores > = 0,2 en el perfil 3D/1D, con lo cual se valida que la estructura propuesta, representada en el modelo 4, homóloga para MerB de P. luorescens, es clasificada de alta calidad y posee coherencia entre la secuencia y la estructura modelada.

Todos los resultados anteriormente presentados en el documento confirman que el modelo planteado, modelo 4, representa una propuesta bastante sólida de la posible estructura 3D para la proteína MerB de P. luorescens (Figura 11).

Figura 11 Modelo final propuesto para la proteína MerB de P. fluorescens. En color rosa se muestran las hélices, en amarillo las hojas beta, cintas azules giros y se muestra el sitio activo: en rojo Cys 99, en azul Asp 102 y en verde Cys 162.
El sitio activo se determinó mediante un alineamiento a partir del molde 3FN8 y la secuencia proteica MerB de P. fluorescens por medio de la aplicación del European Bioinformatics Institute [21], T-Coffe [22].
Discusión
En la literatura estudiada, se ha identificado y estructurado la proteína organomercurial liasa (MerB), de la cual no se encuentra una estructura única, puesto que se ha presentado en cada microorganismo con variantes genómicas, encontrándose entre ellos dos arqueas y 11 filos bacterianos [35]. Además, aún no se han reportado estructuras determinadas experimentalmente ni modelamientos de esta proteína, para la bacteria estudiada en el presente artículo, P. fluorescens. Por esta razón, se inició el trabajo desde su secuencia genómica, con el fin de predecir, por medio de la estrategia de modelamiento por homología con técnicas bioinformáticas, un modelo de estructura proteica de esta molécula. Se identificó que la secuencia de MerB de P. fluorescens la constituyen 212aa, de igual manera y con la ayuda del programa Geno3D se estableció que la estructura con mayor similitud a la molécula con la que trabajamos corresponde a la MerB en complejo con mercurio de Escherichia coli (código PDB: 3FN8), ya que fue la que presentó el valor E más bajo (E = 2 x 10-50), por lo que esta fue usada como molde para obtener un modelo presentado.
Mediante el estudio de la estructura proteica de los modelos ya existentes, se identificó la región característica de este tipo de proteínas biorremediadoras de mercurio, definido como el complejo metálico con mercurio. Y es, precisamente, en esta región, su sitio activo, donde surgen las mayores variantes entre las proteínas MerB de cada microorganismo [9]. Sin embargo, es de resaltar que en los estudios de similitud de las proteínas MerB han demostrado ser homólogas entre ellas [14]. En el análisis del alineamiento, todas las secuencias de Pseudomonas sp analizadas para la familia MerB mostraron un 91% de posiciones conservadas.
En nuestro estudio, se identificó que el sitio activo de la familia de las proteínas MerB está conformado, en su mayoría, por un ion mercurio unido a una triada de aminoácidos. En el caso 3FN8 (molde utilizado), posee un ion de mercurio (Hg+2), el cual, por lo regular, forma una triada catalítica con dos residuos de cisteínas (Cys 96 y Cys 159). En su mayoría son conservadas en las secuencias conocidas hasta el momento y que a su vez trabajan con un residuo de ácido aspártico (Asp 99), donde este, por el contrario, es generalmente variante; de esta manera, la indicada en 3FN8 es la variante más presente en las MerB de Pseudomona spp, pues se han encontrado en otras moléculas uniones en este punto con residuos de serina o cisteína, encontrada en pocas variantes [15].
Por otra parte, en estudios anteriores se ha visto que MerB posee un modelo nativo con sitio activo de solo cisteínas, donde evolutivamente se han reportado mutaciones como las ya mencionadas, pero con una actividad catalítica moderadamente más baja; aquella MerB es de solo cisternas pertenecientes a E. coli, la cual presentó más amplio espectro en compuestos organomercuriales y Hg (II), lo cual no confirmaban que pasaba lo mismo con Pseudomona [36, 37]. Otro reporte que confirma la diferencia de actividad catalítica es la detección de la esencialidad de las cisteínas presentes, siendo la C159 la principal en la catálisis y la C160 con menos efectividad; se recuerda que es en este último aminoácido que P. fluorescens posee un cambio de aminoácido. Por tanto, es en este punto y la razón por la cual es donde se presentan las mutaciones. Sin embargo, la ausencia de cisterna afecta la función catalítica de MerB, puesto que C159/C160 son las más expuestas y flexibles en su estructura, y tienen una función similar al par de cisternas expuestas en MerA; tener un par de cisteínas exteriores flexibles contribuyen a desplazar los compuestos organomercuriales exógenos, logrando una amplia aceptación del sustrato [14].
En el caso de la molécula modelada, el sitio activo del modelo 4 se identificó mediante un alineamiento por pares a través de la aplicación T-Coffe [22] (Figura 12) entre 3FN8 (molde) y la secuencia de P. fluorescens estudiada.

Figura 12 Alineamiento por pares mediante T-Coffee entre 3FN8 (molde) y la secuencia de P. fluorescens estudiada (con rojo Cys 99, azul Asp 102 y verde Cys 162).
Mediante esta estrategia, se determinó que para el caso de la secuencia a partir de la cual se realizó el modelado, esta presenta exactamente los mismos residuos en las posiciones antes descritas (dos cisteínas y un ácido aspártico). Sin embargo, en el caso del modelo presentado, estas no corresponden exactamente a la ubicación, ya que para su alineamiento fue necesario introducir tres espacios en la secuencia de 3FN8 (molde usado), corriendo exactamente cada aminoácido del sitio activo de P. fluorescens tres aminoácidos a su derecha; por tanto, su sitio activo es Cys 99, Asp 102 y Cys 162.
La razón de este cambio de ubicación puede deberse a una razón evolutiva, como se dijo en la presentación de 3FN8; esta proteína fue aislada de E. coli, donde evolutivamente se presenta después de P. fluorescens (Figura 13). Por tanto, se postula que a través del tiempo debido a las mutaciones, E. coli sufrió deleciones en su secuencia, mientras que los genomas de las especies de P. fluorescens aún poseen aquellas regiones como aminoácidos conservados.

Figura 13 Árbol filogenético de bacterias. Se señala que el género Pseudomona spp apareció evolutivamente primero que E. coli. Tomada y editada de [38].
Además, las mutaciones en este tipo de microorganismos biorremediadores se presentan frecuentemente, demostrándose desde su comienzo, pues los linajes de evolución temprana tienden a presentar solo MerA; pues gracias a las presiones ambientales que ocasionan la evolución, el operón Mer logró reclutar la proteína MerB, la cual hace que los linajes de evolución más recientes presenten ambos precursores. Lo anterior se evidencia desde hace poco tiempo; en la actualidad, hay minoría en homólogos de MerB y pocos estudios en su estructura proteica. Gracias a sus ancestros se logró este cambio, donde MerB tiene su ancestro en un mesófilo y MerA en un termófilo, lo cual hizo que desde ese momento existiera más homólogos mesófilos y no solo termófilos, dando un punto positivo para la biorremediación, logrando que microorganismos como E. coli se encuentre dentro de este tipo de linaje mutando con respecto a P. fluorescens [35].
Pero tal cambio de ubicación no afecta en su mecanismo de acción, donde las cisteínas presentes en el sitio activo poseen la función en la unión del sustrato, la ruptura del enlace carbono-mercurio y la regulación de la liberación de iones de mercurio desde el sitio activo; mientras que su otro aminoácido cumple una función complementaria o si se encuentra una cisterna, esta poseería una función estructural [39], o en este caso si se encuentra el ácido aspártico funciona como un donante de protón [40], ya que después de la escisión química de ligandos nucleofílicos de carbono-mercurio, teniendo alta actividad C-99, es necesario posteriormente la protonólisis del enlace carbono-mercurio, donde se postula que el residuo D-102 desprotonaría a C-99, pues el carboxilato D-102 está más próximo a C-99 (3,6 A) en forma libre que C-162 (4,3 A) [36]. Tal mecanismo de acción también se lleva a cabo gracias a la estructura de este mismo sitio activo de MerB, descrito como "sitio activo enterrado", un proceso llamado "canalización del sustrato", y es una alternativa a un mecanismo disociativo, ya que en el primer paso donde MerB desprotoniza, se libera el ion mercurio siendo aún más tóxico que el sustrato, el cual es altamente tóxico para las demás células; pero es esta hendidura la que evita tal afectación, pues desplaza al mercurio iónico a MerA para seguir con su reducción y evitar que proceda al citosol [36].
A partir de comparaciones por homología con diferentes secuencias de MerB, se ha establecido, además, que las proteínas MerB no poseen alta especificidad en el reconocimiento del sustrato, puesto que a partir de las secuencias de MerB de bacterias resistentes a organomercuriales aisladas, se evidenció que el ácido aspártico presenta mayor actividad de escindir enlaces C-Hg en comparación con otro residuo como lo es la serina [36]. Según estudios, las proteínas MerB con un residuo de serina están plegadas incorrectamente y no cortan los enlaces carbono-mercurio en las mismas condiciones que MerB [15].
Estructuralmente, el modelo postulado posee una composición estable y se identifica, por medio de la aplicación PSIPRED, una estructura secundaria de nueve tramos de aminoácidos con tendencia a formar láminas beta y siete tramos con tendencia a hélices alfa, unidos a su vez con pequeños giros, los cuales pueden ser confirmados en RasMol (Figura 5); y mediante el gráfico de Ramachandran, se clasifican en una estructura "tradicional", que favorece su homología hacia las proteínas nativas de su familia y logra una estructura totalmente sin elementos problemáticos o erróneos en el modelo; se evidencia en los puntajes de calidad en ProsaWeb, por lo que se sugiere que el modelo propuesto es coherente entre su secuencia y su modelo tridimensional.
Conclusiones
El modelamiento por homología se reconoce como estrategia útil cuando hay carencia en información experimental reportada en bases de datos y se necesite realizar un ensayo experimental relacionado con la estructura y el funcionamiento de una proteína. Las metodologías existentes para la estructuración de proteínas requieren de alta precisión, ya sea mediante la difracción de rayos X o por resonancia magnética nuclear (RMN), abriendo un campo de investigación en la química computacional. Los resultados del análisis bioinformático de modelamiento por homología indican que la proteína MerB de P. fluorescens es homóloga con la enzima MerB en complejo con mercurio de Escherichia coli 3FN8, el cual es el molde para postular un modelo preciso, el modelo 4. Tales resultados relacionan a la proteína MerB de P. fluorescens como biorremediadora de mercurio, aportando conocimiento en el mecanismo de resistencia a metales más estudiados en la biorremediación. Tales resultados computacionales dan a conocer una plataforma para implementar más estudios y proporcionar referencias sobre el potencial biotecnológico de esta molécula, para el tratamiento de toda fuente contaminada por mercurio a través de sistemas biológicos.

