










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Biológica Colombiana
Print version ISSN 0120-548X
Acta biol.Colomb. vol.16 no.1 Bogotá Jan./Apr. 2011
ENFERMEDAD DE HUNTINGTON: MODELOS EXPERIMENTALES Y PERSPECTIVAS TERAPÉUTICAS
Huntington'disease : Experimentals Models and Therapeutic Perspectives
TERESA SERRANO SÁNCHEZ1, Dra.; LISETTE BLANCO LEZCANO1, DrC; ROCÍO GARCÍA MINET1, M.Sc.; ESTEBAN ALBERTI AMADOR1, DrC; IVÁN DÍAZ ARMESTO2, Dr.; NANCY PAVÓN FUENTE1, DrC; LOURDES LORIGADOS PEDRE1, DrC; MARÍA ELENA GONZÁLEZ FRAGUELA1, Dra.; JORGE FELIPE MONTERO LEÓN3, Dr.; LISIS MARTÍNEZ MARTÍ1, M.Sc.; MARÍA DE LOS ANGELES ROBINSON AGRAMONTE1, DrC; LILIANA FRANCIS TURNER4, DrC. 1 Centro Internacional de Restauración Neurológica. Avenida 25 # 15805 entre 158 y 160. Cubanacán, Playa. Código Postal 11300. Cuidad de la Habana, Cuba. 2 Hospital Clínico Quirúrgico -Juaquín Albarrán-. Avenida 26. Playa. Cuidad de la Habana, Cuba. 3 Instituto Nacional de Oncología y Rehabilitación (INOR). 29 y E. Vedado. Cuidad de la Habana, Cuba. 4 Universidad del Tolima. Ibagué, Tolima, Colombia. Correspondencia a: Teresa Serrano Sánchez, Centro Internacional de Restauración Neurológica. Avenida 25 # 15805 entre 158 y 160. Cubanacán, Playa. Código Postal 11300. Cuidad de la Habana, Cuba. Fax: (537) 33 6339 - 33 2420 - 33 6302. Telefax: 33 6028. Teléfonos del CIREN: 271 5353 - 271 5379. teresa.serrano@infomed.sld.cu
Presentado 9 de septiembre de 2010, aceptado 11 de febrero de 2011, correcciones 22 de febrero de 2011.
RESUMEN
La enfermedad de Huntington (EH) es un trastorno degenerativo de Weiss de origen hereditario. Hasta el momento no existe un tratamiento efectivo para la enfermedad que inexorablemente después de transcurridos 15 a 20 años, evoluciona hacia incapacidad total o muerte. En este trabajo se revisan las características clínicas y morfológicas de la EH y los modelos experimentales más utilizados para su estudio tomando como fuente, artículos indexados en la base de datos Medline publicados en los últimos 20 años. Se valoran las ventajas y desventajas de estos modelos y su perspectiva para el desarrollo de ensayos clínicos. El consenso de lo reportado plantea que de los modelos tóxicos, los inducidos por neurotoxinas tales como ácido quinolínico parecen ser los más adecuados para reproducir las características neuropatológicas, y por otro lado los modelos genéticos contribuyen con más evidencias al conocimiento del origen etiológico de la enfermedad. Numerosos tratamientos han sido aplicados en el manejo de las manifestaciones clínicas que aparecen en EH, sin poder detener o disminuir las afectaciones que derivan de la pérdida neuronal. La sintomatología clínica ha sido posible reproducirla, al menos en parte, en animales de experimentación lo que ha permitido realizar ensayos terapéuticos. Desde el punto de vista de tratamiento, lo que más promisorio parece ser, la terapia celular con células provenientes de diferentes fuentes y dentro de ellas las no neurales, que implican menor censura ética y mayor factibilidad de obtención para la aplicación en los enfermos. Por otro lado el desarrollo de la tecnología del ARN de interferencia, emerge como una herramienta terapéutica potencial para el tratamiento de EH, así como para responder interrogantes básicas relacionadas con el desarrollo de la enfermedad.
Palabras clave: Enfermedad de Huntington, corteza, estriado, modelos experimentales.
ABSTRACT
Huntington'disease (HD) is a degenerative dysfunction of hereditary origin. Up to date there is not, an effective treatment to the disease which having lapsed 15 or 20 years advances inexorably, in a slow form, toward the total inability or death. This paper reviews the clinical and morphological characteristics of Huntington's disease as well as the experimental models more commonly used t study this disease, having as source the articles indexed in Medline data base, published in the last 20 years. Advantages and disadvantages of all experimental models to reproduce the disease as well as the perspectives to therapeutic assay have been also considered. The consent of outline reported about the toxic models, those induced by neurotoxins such as quinolinic acid, appears to be the most appropiate to reproduce the neuropathologic characteristic of the disease, an genetic models contributing with more evidence to the knowledge of the disease ethiology. Numerous treatments ameliorate clinical manifestations, but none of them has been able to stop or diminish the affectations derived from neuronal loss. At present time it is possible to reproduce, at least partially, the characteristics of the disease in experimentation animals that allow therapy evaluation in HD. From the treatment view point, the more promissory seems to be transplantation of no neuronal cells, taking into account ethical issues and factibility. On the other hand the new technology of interference RNA, emerges as a potential therapeutic tool for treatment in HD, and to respond basic questions on the development of the disease.
Key words: Huntington disease, cortex, striatum, animal models.
INTRODUCCIÓN
La enfermedad de Huntington (EH), es una enfermedad neurodegenerativa autosómica dominante que fue descrita por George Huntington en 1872 (Huntington, 1909). El comienzo de la enfermedad es lento, se sitúa en la edad media de la vida, habitualmente entre los 30 y 50 años. Se manisfiesta por movimientos musculares anormales, entre los cuales, el más frecuente, pero no el único, es corea, y pérdida progresiva de funciones cognitivas (García de Yébenes et al., 1992). Los datos epidemiológicos de la EH se obtienen a través del estudio de familias afectadas, y se ha demostrado por estudios de prevalencia el origen noreuropeo de la mutación que conduce a la EH destacándose una densidad de incidencia que va desde 2,5 a 9,95 por cada 100.000 habitantes, lo que indica una prevalencia diferente según la situación geográfica (Harper, 1996). Tomando como fuente, artículos indexados en la base de datos Medline publicados en los últimos 20 años, pudimos constatar que desde el punto de vista neuropatológico, hay muerte celular que afecta principalmente al estriado y corteza cerebral. Neuroquímicamente en el estriado de pacientes con EH hay disminución en la concentración de los neurotrasmisores glutamato y ácido-�-amino butírico (GABA; Dure et al., 1991). La causa molecular de la enfermedad es una expansión del codón CAG, en el primer exón, que codifica para una cadena de glutamina de la proteína denominada huntingtina, cuya función es aún desconocida. La expansión de poliglutamina representa una causa importante de neurodegeneración, ya que resulta ser responsable de las alteraciones observadas en otras enfermedades hereditarias, además de EH (Burright et al., 1997).
Los síntomas iniciales se manisfiestan con frecuencia por un cambio de la personalidad, pero los movimientos coreicos pueden ser el primer signo de la enfermedad (Marsden, 1993). Los trastornos mentales iniciales a menudo son sutiles, aunque se pueden encontrar alteraciones de sensibilidad, de conducta e irritabilidad, y tendencia al comportamiento agresivo o sexual no controlado (Buller et al., 1994). Son frecuentes la depresión y el suicidio entre estos pacientes siempre constituye un riesgo. A medida que la enfermedad progresa, los movimientos coreicos son más intensos y grotescos. Con frecuencia aparece demencia que con el tiempo se vuelve más pronunciada (Marsden, 1993; Buller et al., 1994; Diamond et al., 1992). La marcha, habla y uso de manos se hallan afectados. Igualmente, muchos pacientes experimentan rigidez y acinesia crecientes, que llevan a reducción de la corea. La muerte se produce alrededor de los 15 y 20 años después del comienzo de la enfermedad (Marsden, 1993) . En general los cuadros clínicos son muy variables (Bruyn y Went, 1986). Los sindromes clínicos más característicos son:
- Variante clásica: Inicio entre los 30 y 50 años. Cuadro típico de trastorno motor, cognitivo y de conducta. Progresión hacia la muerte en 20 años.
- Variante senil: Inicio después de los 55 años. Trastorno motor con predominio de corea sin deterioro intelectual ni acortamiento de la vida.
- Variante juvenil: Inicio antes de los 20 años. Sindrome acinético rígido, con grave deterioro mental y muerte en menos de 15 años.
EL ESTRIADO FORMA PARTE DE LOS GANGLIOS BASALES (GB)
En la Fig. 1 se presenta un esquema general del sistema motor, que incluye el lugar que corresponde a los GB. El Estriado (St; caudado y putamen) es una de las estructuras que se encuentra afectada en EH y que forma parte de los GB, es el núcleo integrador y principal receptor de las conexiones más importantes provenientes de la corteza cerebral (Barnes, 1983; Alexander y Crutcher, 1990; Graybiel, 1990; DeLong, 1990). Está formado por diferentes tipos de células neurales, pero las neuronas espinosas de tamaño mediano, que utilizan como neurotransmisor GABA (neuronas gabaérgicas), representan el 70 % de las neuronas encontradas y son sus principales células de proyección (Alexander y Crutcher, 1990; Scortticati y Micheli, 1998). El resto de los tipos neuronales del St son menos frecuentes e incluyen neuronas con dendritas largas que utilizan como neurotransmisor acetilcolina y que funcionan como interneuronas. En EH, las neuronas que sufren mayor afectación, son las neuronas espinosas de tamaño mediano localizadas en esta estructura.

VISIÓN ACTUAL DEL CIRCUITO MOTOR
El concepto clásico de circuito motor corticosubcortical vertical se ha visto modificado en los últimos años por la aparición de otras vías anatómicas que intervienen en el control motor. En la actualidad en el control motor se habla de la existencia de circuitos -transversales-, que se establecen entre los núcleos integrantes de los GB ó desde el tálamo permitiendo la autorregulación de la actividad neuronal antes de proyectar de nuevo a la corteza motora. Estos circuitos incluyen:
- a.
- Circuito Globo pálido externo (GPe)-núcleo subtalámico (NST)-Globo pálido interno (GPi): En el existen aferencias gabaérgicas del GPe al GPi y aferencias glutamatérgicas del NST al GPe, ellas describen un triángulo que permite la autorregulación interna de la actividad de los tres núcleos.
- b.
- Circuito GPi-núcleo centromediano parafascicular del tálamo (CM/Pf)-estriado (STR): El núcleo CM/Pf recibe aferencias desde el GPi y proyecta al STR y al NST desde donde se envían eferencias al GPi con carácter funcional opuesto (GABAérgica y glutamatérgica respectivamente), estableciéndose así un sistema de regulación.
Y aunque no se define como un circuito transversal se plantea que existen conexiones recíprocas del NPP (núcleo pedúnculo pontino), con todos los núcleos de los GB y de modo relevante con el NST y el GPi, estableciéndose así otro sistema de regulación (Rodriguez et al., 2004).
BASES NEURALES DE LAS ALTERACIONES MOTORAS EN LA EH
Las neuronas espinosas (NE) de tamaño medio son las más afectadas. La proyección de estas neuronas se efectúa a través de dos vías, denominadas directa e indirecta (Gerfen, 1992). En la primera, éstas neuronas del estriado portadoras de sustancia P y GABA ejercerían su acción inhibitoria sobre neuronas del pálido interno y las de la pars reticulata, de la SN (con GABA como neurotrasmisor principal), permitiendo así la acción del tálamo sobre la corteza. Al perderse la acción inhibitoria de estas NE sobre el pálido interno y SN pars reticulata, estos núcleos podrían ejercer la suya libremente sobre el tálamo. Se presupone que este mecanismo conduciría así a la bradicinesia, que se observa en algunos casos de EH (Gerfen, 1992). La vía indirecta implicaría a un circuito algo más complejo. Las NE encefalínicas proyectan desde el estriado su acción inhibidora sobre otro núcleo inhibidor, el pálido externo, cuya activación inhibe al subtálamo, este núcleo glutamatérgico estimula al pálido interno y SN pars reticulata, permitiéndoles ejercer su acción inhibidora sobre el tálamo. Se supone que en la EH se produciría una situación inversa, la pérdida de las NE impediría ejercer la acción inhibitoria del estriado sobre el pálido externo, el cual podría inhibir libremente al subtálamo, este dejaría así de estimular al pálido interno y SN pars reticulata, quedando el tálamo libre de esta influencia inhibitoria, apareciendo entonces corea (Reinier et al., 1998; Storey y Beal, 1993).
BASES MOLECULARES Y PATOGÉNICAS DE LA EH
En el año 1993 el grupo de investigación colaborativo de la enfermedad de Huntington (The Huntigton' Disease Collaborative Research Group, 1993) descubrió el origen genético de la enfermedad la cual es autosómica dominante con penetrancia completa, encontrándose asociada a una repetición del triplete de nucleótidos CAG en la región que codifica el gen de huntingtina (Laird, 1990). En la población normal los trinucleótidos de CAG están repetidos en un número menor de 30 repeticiones. En el caso de los enfermos, existe un número de repeticiones mayor de 36 llegando a hacer en algunos casos superiores a 100 (McDonald y Gusella, 1996). Esta mutación consiste en la inserción de múltiples copias de dicho trinucleótido en diferentes regiones de un gen localizado en el brazo corto del cuarto par de cromosomas, específicamente en el gen IT15. Este cromosoma es el encargado de codificar la proteína huntingtina la cual se expresa no sólo en cerebro sino también en diferentes tejidos. Debido a la mutación genética, en los pacientes con EH la huntingtina es sintetizada con un exceso de glutamina, lo cual contribuye a que la misma se agregue formando complejos insolubles con ubiquitina y otras proteínas fibrilares, apareciendo de manera característica, como cuerpos de inclusión en el núcleo (McDonald y Gusella, 1996; Imarisio et al., 2008). Aunque su función permanece desconocida se sabe que su unión a estas proteínas pudiera estar asociada a la función con la cual ellas están relacionadas: trasmisión de señales neuronales y regulación de la muerte neuronal apoptótica (Alexi et al., 2000; Beal et al., 1994a). Se ha reportado que el depósito de esta proteína anormal provoca en etapas tempranas de la enfermedad, muerte de neuronas de proyección gabaérgicas espinosas de talla media que expresan encefalinas (Kowall et al., 1987).
El descubrimiento de esta proteína ha abierto una nueva perspectiva en la patogenia y tratamiento de la EH, aunque los mecanismos responsables de la muerte celular selectiva y pérdida de neuronas no son del todo conocidos hasta el momento (The Huntigton' Disease Collaborative Research Group, 1993; Imarisio et al., 2008).
Los principales hallazgos neuropatológicos de la EH se encuentran localizados en el estriado, donde se ha evidenciado pérdida de neuronas de proyección, unido a una preservación de interneuronas espinales grandes (Reinier et al., 1998).
Para explicar la patogenia de la EH han sido implicados diversos mecanismos: excitotóxicos, metabólicos y de estrés oxidativo (Tkac et al., 2001; Browne, 1999).
El daño excitotóxico es producido por la sobreactividad local de aminoácidos excitatorios (AAE) sobre sus receptores, lo cual conduce a despolarización sostenida que provoca muerte celular selectiva. Glutamato u otros AAE endógenos que actúan sobre varios tipos de receptores glutamatérgicos, pudieran estar involucrados en la patogenia de la EH. (Spencer et al., 1987; Biscoe et al., 1975). Estos receptores glutamatérgicos son de dos tipos: metabotrópicos y ionotrópicos. Los metabotrópicos son aquellos que se encuentran ligados a proteínas G e incluyen L-AP4 (ácido L-2-amino-4-fosforopro-piónico) y los ionotrópicos son canales iónicos e incluyen a los NMDA (N-metil-D-aspartato), los kaínato y los AMPA (ácido-amino-3-hidroxi-5-metil-4-isoxasolpropiónico; Nakanishi, 1992; Schoepp y Con, 1993). Aunque todos pueden mediar muerte celular, tanto apoptótica como necrótica, desencadenada por mecanismos excitotóxicos, son los del tipo NMDA los que tienen una acción más relevante, y los que se encuentran más ampliamente distribuidos en el cerebro (Coyle y Puttfarcken, 1993; Giffard et al., 1992).
La neurotoxicidad por aminoácidos excitatorios, no depende solamente de los niveles endógenos de los posibles compuestos neurotóxicos, ni del grado de eficacia de sus mecanismos de re-captación, sino también de otros factores que modulan la respuesta, y por tanto la actividad neuronal a nivel del receptor o post-receptor. A nivel del receptor se han descrito compuestos endógenos que inhiben de forma competitiva el receptor NMDA (N-Metil-D-Aspartato) y sustancias que lo modifican alostéricamente aumentando su afinidad por su ligando (Mayer et al., 1984; Wong et al., 1986; Lodge y Golingridge, 1990; Kemp et al., 1988). A nivel post-receptor, el efecto de aminoácidos excitatorios puede ser modificado por manipuladores de canales iónicos, segundos mediadores, activadores o inhibidores de proteínas quinasas, factores de crecimiento y otros fármacos (Novelli et al., 1988; Weiss et al., 1990).
Estudios en cultivo de tejido indican que la muerte neuronal mediada por receptores glutamatérgicos, puede seguir dos vías diferentes: una forma aguda y una retardada (Beal et al., 1994a; Beal, 1996). La neurotoxicidad aguda se caracteriza por una dilatación de la célula en presencia de agonistas glutametérgicos, que trae como consecuencia la lisis osmótica de la neurona (necrosis). Los dos iones responsables de la entrada masiva de agua en este caso son Na+ y Cl-. La degeneración neuronal retardada es mediada por mecanismos dependientes de Ca2+. Este puede mediar muerte celular por varios mecanismos que incluyen activación de proteínas quinasas, fosfolipasas A2, óxido nítrico sintetasa, proteasas, generación de radicales libres, daño mitocondrial e inhibición de la síntesis de proteínas (Coyle y Puttfarcken, 1993; Giffard et al., 1992; Beal, 1996; Steller, 1995; Mitchell et al., 1994; Quintanilla y Johnson, 2009). La inyección intraestriatal de agonistas de estos receptores reproduce muchas de las características neuroquímicas y neuropatológicas de la EH.
Con relación a los mecanismos metabólicos planteados en la patogenia de la EH, existe una hipótesis en la que se aúnan el conocimiento de los mecanismos excitotóxicos mediados por receptores NMDA con defectos en el metabolismo energético detectados en la EH (Beal, 1992; Oliveira, 2010). Según esta hipótesis los mecanismos excitatorios contribuirían a muerte neuronal de células con dificultades metabólicas.
Relacionado con estrés oxidativo se plantea que la cadena respiratoria mitocondrial genera radicales libres que alteran la función celular normal. El óxido nítrico producido por la óxido nítrico sintetasa, se ha detectado en interneuronas no espinosas de talla media a grande del estriado, donde actúa como neurotrasmisor. Además puede generar radicales hidroxilo a partir del anión superóxido (Deckel, 2001; Tasset et al., 2009). Estos datos ponen de manifiesto que el estrés oxidativo, tiene un papel relevante en el curso y evolución de la EH, asociándose a distintos estadios de la enfermedad, por lo que puede utilizarse como marcador-pronóstico y de la efectividad de tratamiento, así como constituir un foco de interés para el desarrollo de nuevas estrategias terapéuticas. Finalmente existe una nueva vía patogénica que involucra al sistema inmunológico del individuo, donde existe una activación anormal de la microglia y macrófagos lo que sugiere una disfunción inmune que juega un papel importante en la patología cerebral (Bjorkqvist et al., 2008)
MODELOS EXPERIMENTALES EN LA EH
Los estudios realizados para aumentar el conocimiento de la etiología de la EH, conjuntamente con la constante búsqueda de alternativas terapéuticas, ha hecho incuestionable la importancia del uso de la experimentación animal. Aunque la EH no se manifiesta en los animales de manera espontánea, se han desarrollado modelos experimentales de la enfermedad que han ayudado a estudiar los mecanismos involucrados en la patogenia de la misma, permitiendo desarrollar estrategias terapéuticas (Ferrante, 2009; Perry et al., 2010). No existe un modelo experimental único que satisfaga completamente los requerimientos de similitud con la EH en cuanto a fenomenología clínica, aspecto histológico y genético. Sin embargo, se sabe desde hace más de dos décadas, que la inyección intracerebral de algunas neurotóxinas producen lesiones estriatales que guardan alguna semejanza con las lesiones histológicas observadas en la EH (DiFiglia, 1990). En general se han descrito dos tipos de modelos homólogos para la EH: tóxicos y trangénicos.
MODELOS TÓXICOS
La muerte neuronal excitotóxica lenta puede producirse como consecuencia de un defecto en el metabolismo oxidativo. Las interrupciones en la síntesis de ATP pueden conducir a despolarización neuronal parcial con activación de receptores NMDA, lo cual provocaría daño neuronal excitotóxico secundario (Palfi et al., 1996; La Fontaine et al., 2000; Beal et al., 1993a).
MODELOS EXPERIMENTALES DE LA EH BASADOS EN EXCITOTOXICIDAD
Se ha descrito que los modelos de excitotoxicidad han sido utilizados con mayor frecuencia. Los mecanismos de excitotoxicidad están basados en la sobreexcitación de las neuronas como resultado de estimulación propagada y continua de receptores a aminoácidos excitadores, lo que produce alteraciones serias en la fisiología de las neuronas, conduciéndolas a muerte celular (Olney et al., 1971). Tóxinas con las que pueden ser producidos estos modelos:

- a.
- Ácido kaínico (AK). Es un derivado de algas, agonista del receptor kaínato (Coyle y Schwarcz, 1976; Mason y Fibriger, 1978). En 1976 Coyle y Schwarcz establecieron el empleo de AK para reproducir las características histológicas, conductuales y bioquímicas de la EH, produciendo una lesión de neuronas gabaérgicas y colinérgicas sin alterar a neuronas dopaminérgicas. La actividad de algunas enzimas como glutamato descarboxilasa (GAD) y colina acetil transferasa (ChAT) disminuyeron de forma similar a lo encontrado en la enfermedad. Sin embargo, a diferencia de la EH, se observó disminución significativa en los niveles de somatostatina y neuropéptido Y (Coyle y Schwarcz, 1976).
- b.
- Ácido iboténico (IBO). Es un análogo rígido del glutamato (Barrer y Dunnett, 1994). En 1990, Hantraye et al., propusieron al IBO como inductor de un modelo experimental de la EH, basado en la toxicidad de este ácido en el hipocampo de rata. Cuando se administró en el cuerpo estriado de bovinos, se observó proliferación astrocítica y pérdida de fibras colinérgicas sin alteración de fibras dopaminérgicas. Hubo disminución de metaencefalinas acompañándose de alteraciones conductuales tales como: corea, distonía y asimetría postural. No obstante, también se observaron diferencias conductuales que limitan la reproducibilidad del modelo, siendo mejor alternativa que el del AK, pero con ciertas limitaciones (Hantraye et al., 1990).
- c.
- Ácido quinolínico (AQ). Este derivado del metabolismo del ácido kinurénico (que proviene de una rama del metabolismo del triptófano), reproduce en parte los cambios bioquímicos y neuropatológicos de la EH, demostrando que el AQ es capaz de inducir lesiones neuronales cuya distribución es mucho más parecida a la que produce el AK en el modelo de EH, ya que afecta a neuronas espinosas respetando relativamente a las no espinosas. Entre los cambios más significativos desde el punto de vista neuroquímico se destacan la reducción severa de las concentraciones de GABA en células estriatales, reducción severa de la actividad GAD y gliosis estriatal (Beal et al., 1986; Beal et al., 1991). Se ha demostrado que después de la administración de AQ en el estriado, se producen niveles de somatostatina, neuropéptido Y, sustancia P, vasopresina, GABA y dopamina muy similares a los que se observan en la EH por lo que ha sido considerado como el mejor modelo experimental. En la Tabla 1, se indican los principales cambios de estas sustancias neuroactivas en la EH y, en los diferentes modelos experimentales de la enfermedad.
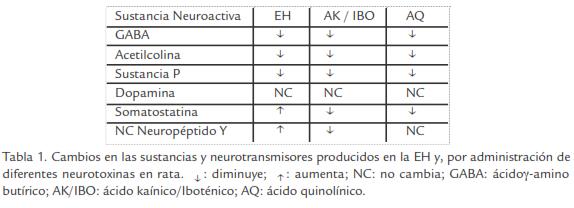
Conductualmente, el AQ produce hiperactividad locomotora y alteraciones del aprendizaje y la memoria. Con este conocimiento, se llegó a proponer al AQ como el principal agente etiológico en la EH, y que las lesiones provocadas por concentraciones submicromolares de esta toxina pueden ser bloqueadas por MK-801, un antagonista no competitivo de receptores NMDA sugiriendo que las alteraciones observadas en los enfermos, podrían estar mediadas por excitación de estos receptores (Albin et al., 1990).
MODELOS EXPERIMENTALES DE LA EH BASADOS EN LA ALTERACIÓN DEL METABOLISMO ENERGÉTICO.
La teoría de la excitotoxicidad explica el mecanismo de muerte neuronal en la EH y en otras enfermedades neurodegenerativas como una primera causa de muerte neuronal,pero una posible explicación de la toxicidad del glutamato podría ser también déficit en la producción de energía mitocondrial (Albin y Greenmyre, 1992; Ankacrona et al., 1995). Por lo tanto se hipotetizó que alteraciones en la fosforilación oxidativa pueden ser el déficit primario que predispone a la degeneración neuronal secundaria a la toxicidad por glutamato (Beal et al., 1993b). Tóxina con la que puede ser producido este tipo de alteración metabólica: Ácido 3-nitropropiónico (3-NP; inhibidor del complejo II de la cadena respiratoria mitocondrial). El mecanismo de daño de esta toxina es la inhibición de succinato deshidrogenasa, enzima que juega un papel central en la cadena de transporte de electrones y en el ciclo de ácidos tricarboxílicos. La administración de dosis bajas de ácido 3-NP produce atrofia estriatal selectiva (Borlongan et al., 1995; Túnez y Santamaría, 2009). Se cree que el daño selectivo causado por 3-NP en ciertas regiones del cerebro está directamente relacionado con la tasa metabólica y la densidad de receptores NMDA de cada región (Hamilton y Gould, 1987).
El modelo del 3-NP ha sido propuesto como una alternativa para producir lesiones similares a las de la EH, ya que por inhibición irreversible del ciclo del ácido cítrico mitocondrial se produce disminución del ATP y elevación de las concentraciones de lactato (Beal et al., 1993a; Gould y Gustine, 1982; Beal, 1994b). Además se han encontrado niveles bajos de glutamato y daño en el metabolismo energético oxidativo como los observados en pacientes con desordenes neurodegenerativos incluyendo EH (Gould y Gustine, 1982; Zorumski y Olney, 1993). Teniendo presente las limitaciones de este modelo (como son el salto filogenético, no expresar la proteína mutada y tener un efecto autolimitado a la biodisponibilidad del tóxico), es importante que este agente es capaz de reproducir, dependiendo de la dosis y del tiempo de suministro, disfunciones motoras, de aprendizaje y lesiones estriatales que mimetizan alteraciones histológicas y neuroquímicas de EH (Túnez y Santamaría, 2009).
Este modelo se ha combinado con el de excitotoxicidad pues se sabe que el efecto neurotóxico del 3-NP es potenciado por agonistas NMDA y disminuido por el pretratamiento con antagonistas de este subtipo de receptor glutamatérgico.
MODELOS GENÉTICOS
Modelos experimentales basados en el defecto genético de la EH. Hay dos categorías de modelos genéticos: trangénicos y knock-in.
Los transgénicos resultan de la inserción al azar en el genoma del ratón, de una porción del gen de huntingtina humana, que contiene repeticiones de tripletes CAG expendidas en el locus IT15, YAC mutante completo para huntingtina etc., todos ellos muy útiles en el estudio de la EH. La expresión de ella puede conducirse por diferentes promotores. En los modelos knock-in, una porción del gen de huntingtina humana se introduce en el locus genético de huntingtina del ratón en el cromosoma 4. El promotor de huntingtina exógeno promueve la expresión y producción de la proteína mutada, de manera espacial y temporal (Kuhn et al., 1995; Heng, 2008; Ramaswamy et al., 2007).
Se ha demostrado que en el estudio del modelo transgénico, en el cual se provoca una expansión de las repeticiones CAG en el locus IT15, se desarrolla un fenotipo de alteraciones neurológicas similares a los síntomas motores observados en la EH (Bates et al., 1997), representando el modelo más cercano de la misma pues su etiología es debida directamente a la misma mutación humana. Sin embargo, de manera sorprendente los efectos del trasplante neural intraestriatal en estos animales no son similares a los observados en los modelos de lesión neurotóxica. Aunque se ha podido constatar que estos trasplantes sobreviven en el estriado hospedero, sus efectos conductuales fueron muy pequeños, particularmente en aquellos animales con deficiencia neurológica profunda (Dunnett et al., 1998).
Los criterios neuropatológicos utilizados para el diagnóstico de la EH son muy complejos e incluyen una secuencia de características clínicas y neuroanatómicas. Los mode-los animales no reproducen en su totalidad toda esta constelación de síntomas naturales o patológicos que aparecen en la enfermedad humana, por lo tanto, la valoración de las similitudes y diferencias que aportan cada uno de estos modelos (Vonsattel, 2008; Ehrnhoefer et al., 2009), nos ayudará a determinar cual de ellos debe ser utilizado en una investigación dada, según la respuesta que deseamos obtener en correspondencia con los objetivos planteados para su interpretación.
Fármaco-terapéutica actual en la EH. No existe tratamiento curativo para estos enfermos. Hasta la fecha todos los tratamientos establecidos se limitan a la mejoría de síntomas que aparecen durante la enfermedad. Un gran número de medicamentos sirven para tratar alteraciones del movimiento y trastornos psicológicos tales como: corea, psi-cosis y depresión. A pesar de que la mayoría de estos medicamentos pueden ser útiles en el manejo de la EH, no existe aún ninguno que detenga o revierta el proceso ya establecido. Estos fármacos se administran en dosis crecientes hasta que se controla la corea. También ayudan a controlar las complicaciones mentales, que con frecuencia son muy angustiantes para las personas que padecen esta enfermedad (Feigin et al., 1995; Kieburtz et al., 1996; Murman et al., 1997).
En la búsqueda de nuevos tratamientos, se ha seguido investigando sobre el comienzo y progresión de la enfermedad, así como el papel de la función mitocondrial y excitotoxicidad, que han permitido llevar a cabo numerosos estudios de terapéutica experimental (Feigin, 1998). Por otro lado, el conocimiento de cambios neuroendocrinos vistos en modelos de roedores, ha dado información de mecanismos críticos, para el desarrollo de nuevas estrategias de tratamiento, especialmente en aquellas intervenciones que se realizan en etapas tempranas de la enfermedad, modificando la progresión natural de esta (Petersén, 2009). La vía patogénica que involucra al sistema inmunológico del individuo, pudiera ser otro blanco terapéutico en la búsqueda de la cura de la enfermedad (Bjorkqvist et al., 2008).
Terapéuticas potenciales para la EH. Teniendo en cuenta los resultados encontrados en la experimentación animal, durante aproximadamente 20 años y la ausencia de una farmacoterapia efectiva, es que se ha iniciado la investigación clínica del trasplante de tejido neural fetal. En 1994 se crea el grupo European Networt for striatal Transplantation in Huntington's Disease (Equivalente al grupo NECTAR para el trasplante en personas con enfermedad de Parkinson), para el trasplante estriatal en personas con EH, y paralelo a esto se desarrolla una prueba de evaluación para dichos pacientes previo a recibir el trasplante (Quinn et al., 1996).
Trasplante de células neurales. En la década de 1980 hubo un incremento en las terapias dirigidas a contrarrestar las alteraciones presentes en etapas tempranas de la EH con el uso de trasplante de tejido neural fetal como un método terapéutico. Esta terapia comenzó mucho más tarde en personas que sufren de EH que en aquellos aquejados de enfermedad de Parkinson. El trasplante de tejido neural fetal constituye una alternativa de tratamiento para la EH, a la que se le ha prestado atención, conjuntamente con el desarrollo de técnicas de cultivo de tejido de células neuronales viables para el trasplante (Kopyoy et al., 1998).
El primer trasplante neural en pacientes con EH fue realizado por Madrazo et al., en 1990 (Madrazo et al., 1991) y en 1991 por Sramba et al., 1992, aunque en este último caso sólo fue experimental, posteriormente esta técnica se mantuvo detenida, para comenzar a ser utilizada nuevamente en un gran número de personas, cuatro años más tarde. Este programa de trasplante neural fue iniciado y seguido por otros países como por ejemplo Francia (Bachoud-Levi et al., 2000), y Estados Unidos (Freeman et al., 2000), donde fueron tratados varios pacientes.
Pudiera resultar muy beneficioso el hecho de poder generar tejido neural humano in vitro. En los momentos actuales se ha establecido que las células madre embrionarias de blastocistos de ratón pueden ser expandidas en cultivo y por tanto lograr que se diferencien en un amplio rango de tejidos, incluyendo células neurales y gliales. En otros estudios se ha podido obtener células madre neurales que han sido aisladas tanto de cerebro de roedores adultos como de aquellos que se encuentran en desarrollo (Svendsen y Smith, 1999) y que han resultado ser apropiadas principalmente en terapias de reemplazo celular (Muller et al., 2006).
A pesar de lo antes expuesto, donde se evidencia la existencia de trabajos que apoyan los efectos positivos de trasplantar células, otros hablan de efectos negativos encontrados a largo plazo en el tejido trasplantado, lo que trae consigo criterios controversiales, de si es beneficioso o no uso de estas células en estos pacientes, como fue demostrado por Cicchetti et al., cuando evidenció la presencia de mecanismos inflamatorios y excitotóxicos que acarrearon degeneración neuronal de los trasplantes realizados en pacientes con HD (Cicchetti et al., 2009), y en el mismo año la demostración por Dirk Keene et al., de la aparición de complicaciones tardías debidas a un crecimiento excesivo en la zona del trasplante que resultó ser crítica para estructuras del sistema nervioso central (Keene et al., 2009).
Opinamos que la consolidación de la terapia celular como tratamiento alternativo en la EH, sigue siendo una esperanza de vida para personas con esta enfermedad ya que tiene como premisa la existencia de una fuente segura de células. En estos momentos las células neurales fetales confrontan importantes cuestiones técnicas y éticas que limitan su obtención. Por esta razón la comunidad científica esta enfrascada en la búsqueda de fuentes celulares alternativas para el trasplante (Fink et al., 2000), células encapsuladas por ingeniería genética que secreten factores tróficos (Emerich, 1997), y fuentes no neurales con estos mismos propósitos. Se ha determinado que estas otras fuentes celulares no nerviosas tienen un alto potencial para el trasplante siendo útiles en el autotrasplante, tanto en terapia génica como celular (Azizi et al., 1999; Kim, 2008; Clelland et al., 2008; Martin et al., 2008)
Trasplante de células no neurales. Existen diversas fuentes de células no neurales que pueden ser utilizadas en el trasplante dentro de ellas se destaca las provenientes de la médula ósea, que han sido utilizadas también en la reparación del daño en el sistema nervioso central (Parr et al., 2007; Scolding et al., 2008).
Potencialidades de las células de medula ósea para ser utilizadas como una fuente alternativa en el trasplante de pacientes con enfermedades neurológicas. La médula ósea adulta (MOA) es de muy fácil acceso y es una fuente rica de células progenitoras y células madre (Prockop, 1997). Un número creciente de las últimas publicaciones han mostrado que células madre de MOA son capaces de generar fenotipo neural tanto in vitro (Sánchez-Ramos et al., 2000; Woodbury et al., 2000) como in vivo después del trasplante (Brazelton et al., 2000; Mezey et al., 2000). Las células derivadas de MOA han sido utilizadas de manera exitosa para el trasplante en animales que han sufrido un daño cerebral traumático (Manhood et al., 2001) y en la isquemia estriatal para reducir el déficit motor que aparece después del daño (Zhao et al., 2002). Más recientemente, se ha utilizado el trasplante de células de MOA autóloga para revertir el déficit cognitivo observado en un modelo animal de EH (Lescaudron, 2003). En este trabajo se pudo demostrar que el trasplante de células de MOA es capaz de reducir el daño cognitivo en este modelo de EH.
En este sentido, nuestra atención se ha enfocado en el uso del trasplante de CMO como estrategia de tratamiento para personas portadoras de esta entidad clínica. Es por ello que nuestros trabajos actuales se han encaminado a evaluar si las CMO al ser trasplantadas dentro del estriado lesionado con AQ son capaces de sobrevivir y modificar las alteraciones conductuales presentes en el modelo de Huntington en ratas.
Estos resultados son prometedores, porque el uso de células madre adulta autóloga minimiza el riesgo de rechazo inmunológico y al mismo tiempo evita problemas éticos que existen con el uso de trasplante de tejido fetal como terapia para enfermedades neurodegenerativas.
Terapia neuroprotectora. La neuroprotección ha sido uno de los pilares explorados en el tratamiento de la EH, donde conociendo el requerimiento trófico de la población neuronal afectada, se puede inducir una respuesta trófica endógena ó a través de la administración exógena del factor neurotrófico específico para prevenir o detener la progresión de la enfermedad (Alberch et al., 2002; Perez-Navarro et al., 2000; Hersch y Rosas, 2008). En la EH, esta acción favorecedora ha sido aportada por BDNF (del inglés brain derived neurotrophic factor), donde se demuestra que esta proteína contribuye de manera sustancial al soporte trófico y supervivencia de neuronas afectadas en esta enfermedad (Ma B et al., 2010).
Terapia génica. La EH es una de las 9 enfermedades neurodegenerativas dominantes que resulta de una expansión de poliglutamina repetida, que cobra una función tóxica (The Huntington's disease colaborative group, 1993), por lo que en esta entidad la terapia génica cobra gran importancia. Hasta el presente se han descrito terapias en las que los animales han sido tratados con sustancias que incrementan trascripción de genes neuroprotectores (Ferrante et al., 2003), prevención de muerte por apoptosis e inhibición de la formación de agregados de poliglutamina (Tanaka et al., 2004; Karpuj et al., 2002), entre otros. Todas estas terapias podrían de manera indirecta afectar la expresión de alelos en la EH. Sin embargo, hasta la fecha no existían terapias, que describieran la reducción directa de la expresión del gen mutado que aparece en la enfermedad. Hoy día se conoce que la terapia que utiliza silenciamiento de la expresión de genes mutados pudiera ser promisoria en la EH, lo cual ya ha sido demostrado en modelos animales en los cuales se provoca silenciamiento de genes mutados que son responsables de las características neuropatológicas y motoras que se observan en la enfermedad, y que han dado el sustento para terapias que utilizan el ARN de interferencia (Yamamoto et al., 2000; Harper et al., 2005; Harper, 2009).
CONCLUSIÓN
Hasta aquí hemos hecho un recorrido por el estado del conocimiento acerca de la EH, específicamente los referidos a las principales manifestaciones clínicas de la enfermedad y los síndromes clínicos que conforman. Se abordaron las estructuras del SNC que se encuentran afectadas, indicando las interrelaciones que existen entre ellas. Se exponen las bases neurales y moleculares que dan origen a esta afectación. Se describen además, los modelos experimentales más utilizados para simular la enfermedad y el ensayo de diferentes terapias sustitutivas y neuroprotectoras. A nuestro juicio esta es una enfermedad de evolución lenta, que por tener carácter hereditario, pudiera ser candidata a estudios encaminados a la búsqueda de marcadores tempranos, indicativos de la enfermedad en sujetos con antecedentes familiares.
La utilidad de uno u otro modelo experimental, dependen de los objetivos que se persigan en la investigación, si tenemos en cuenta que ninguno de los modelos reproduce todas las características morfológicas, cognitivas y conductuales que aparecen en la enfermedad.
Con el avance de las nuevas tecnologías, se han implementado formas de tratamiento que además de aliviar síntomas, contribuyen a sustituir células dañadas y/o restaurar el daño, entre otras vías por efecto trófico sobre el tejido lesionado. Nosotros consideramos que este es un camino prometedor pues de esta manera las personas con dicha afección lograrían alcanzar una mayor calidad de vida.
AGRADECIMIENTOS
Los autores agradecen a todos los contribuidores de este trabajo.
BIBLIOGRAFÍA
ALBERCH J, PEREZ-NAVARRO E, CANALS JM. Neuroprotection by neurotrophins and GDNF family members in the excitotoxic model of Huntington's disease. Brain Res Bull. 2002;57:817-822.
[ Links ]ALBIN RL, YOUNG AB, PENNEY LB, HANDELIN B, BALFOUR R. Anormalities of striatal projection neurons and N-methyl-D-aspartate receptors in presymtomatic Huntington's disease. N Engl J Med. 1990;322:1293-1298.
[ Links ]ALBIN RL, GREENAMYRE JT. Alternative excitotoxic hypothesis. Neurology. 1992;42:33-738. [ Links ]
ALEXANDER GE, CRUTCHER MD. Functional architecture of Basal Ganglia circuit: Neural substrates of parallel processing. Trends Neurosci. 1990;13:266-271. [ Links ]
ALEXI T, BORLONGAN CV, FAULL RL, WILLIAMS CE, CLARK RG, GLUCKMAN PD, et al. Neuroprotective strategies for basal ganglia degeneration: Parkinson' and Huntington's diseases. Prog Neurobiol. 2000;60:409-470.
[ Links ]ANKACRONA M, DYPBUKT JM, BONFOCO E, ZHIVOTOVSKY B, ORRENIUS S, LIPTON SA. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961-973. [ Links ]
AZIZI SA, STOKES D, AUGELLI BJ, DIGIROLAMO C, PROCKOP DJ. Engraftment and migration of human marrow stromal cell implanted in the brains of albino rats- similarities to astrocytes grafts. Hum Gene Ther. 1999;10:2539-2549. [ Links ]
BACHOUD-LEVI A, BOURDET C, BRUJIERES PJ, NGUYEN P, GRANDMOUGIN T, HADDAD B, et al. Safety and tolerability assessment of intrastriatal neural allografts in five patients with Huntington's Disease. Exp Neurol. 2000;161:194-202. [ Links ]
BARKER RA, DUNNETT SB. Ibotenic acid lesions of the striatum reduce drug-induced rotation in the 6-hydroxydopamine-lesioned rat. Exp Brain Res. 1994;101:365-374. [ Links ]
BARNES CD. The basal ganglia in extrapyramidal dysfunction. Brain Res Bull. 1983;11:271-275. [ Links ]
BATES GP, MANGIARINI L, MAHAL A, DAVIES SW. Transgenic Models of Huntington'Disease. Hum Mol Genet. 1997;6:1633-1637. [ Links ]
BEAL MF, KOWALL NW, ELLISON DW, MASUREK MF, SWARTZ KJ, MARTIN JB. Replication of the neurochimical characteristics of Huntington's disease by quinolinic acid. Nature. 1986;321:168-171. [ Links ]
BEAL MF, FERRANTE RJ, SWARTZ KJ, KOWALL NW. Chronic quinolinic acid lesions in rats closely resemble Huntington's disease. J Neurosci. 1991;11:1649-1659. [ Links ]
BEAL MF. Does impairment of energy metabolism result in excitotoxic neuronal death in neurodegenerative illnesses? Ann Neurol. 1992;31:119-130. [ Links ]
BEAL MF, BROUILLET E, JENKINS B, FERRANTE RJ, KOWALL NW, MILLER JM, et al. Neurochemical and histologic caracterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci. 1993a;13:1481-1492. [ Links ]
BEAL MF, BROUILLET EP, JENKINS B, HENSHAW R, ROSEN B, HYMAN BT. Age-dependent striatal excitotoxic lesions produced by the endogenous mitochondrial inhibitor malonate. J Neurochem. 1993b;61:1147-1150. [ Links ]
BEAL MF, JOLLES G, STUTZMANN JM. Huntington's disease. En: Jolles G., Stutzmann J.M., editores. Neurodegenerative diseases. London: Academic Press; 1994a:169-181. [ Links ]
BEAL MF. Neurochemistry and toxin models in Huntington's disease. Curr Opin Neurol. 1994b;7:542-547. [ Links ]
BEAL MF. Mechanisms of excitotoxicity in neurological disease. FASEB J. 1996;6:3338-3340. [ Links ]
BISCOE TJ, EVAS RH, HEADLEY PM, MARTIN MR, WATKINS JC. Domoic acid and quisqualic acid as a potent amino acid excitants of frog and rat spinal neurons. Nature. 1975;255:166-167. [ Links ]
BJORKQVIST M, WILD EJ, THIELE J, SILVESTRONI A, ANDRE R, LAHIRI N, et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington's disease. Jpn J Exp Med. 2008;205:1869-1877 [ Links ]
BORLONGAN CV, KOUTOUZIS TK, FREEMAN TB, CAHILL DW, SANBERG PR. Behavioral pathology induced by repeated systemic injections of 3-nitropropionic acid mimics the motoric symptoms of Huntington's disease. Brain Res. 1995;697:254-257. [ Links ]
BRAZELTON TR, FABIO M, ROSSI V, KESHET GL, BLAU HM. From marrow to brain: Expression of Neuronal Phenotypes in adult mice. Science. 2000;290:1775-1779. [ Links ]
BROWNE SE, FERRATE RJ, BEAL MF. Oxidative stress in Huntington's disease. Brain Pathol. 1999;9:147-163. [ Links ]
BULLER N, SALMON D, HEINDEL WC. Specificity of the memory deficit associated with basal ganglia dysfunction. Res Neurol. 1994;150:560-567. [ Links ]
BRUYN GW, WENT LN. Huntington's Chorea. En Ed. Vinken PJ Bruyn GW, Klawans, Handbook of Clinical Neurology. Extrapiramidal Disorders. Amsterdam, Elsevier Science Publishers; 1986;5:267-297. [ Links ]
BURRIGHT EN, ORR HT, CLARK HB. Mouse models of human CAG repeat dosorders. Brain Pathol. 1997;7:965-977.
[ Links ]CICCHETTI F, SAPORTA S, HAUSER RA, PARENT M, SAINT-PIERRE M, et al. Neural transplants in patients with Huntington's disease undergo disease-like neuronal degeneration. Proc Natl Acad Sci U S A. 2009;8:12483-12488.
[ Links ]CLELLAND CD, BARKER RA, WATTS C. Cell therapy in Huntington disease. Neurosurg Focus. 2008;24:1-12.
[ Links ]COYLE JT, SCHWARCZ R. Lesions of striatal neurones whith kainic acid provides a model for Huntinton' chorea. Nature. 1976;263:244-246.
[ Links ]COYLE JT, PUTTFARCKEN P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262:689-695.
[ Links ]DECKEL AW. Nitric oxide and nitric oxide synthase in Huntington's disease. J Neurosci Res. 2001;64:99-107.
[ Links ]DELONG MR. Primate Models of movement disorders of basal ganglia origin. Trends Neurosci. 1990;13:281-285.
[ Links ]DIAMOND R, WHILE RF, MYER RH, MASTROMAURO C, KOROSHEU WJ, BULLER N, et al. Evidence of presymptomatic cognitive decline in Huntington's disease. J Clin Exp Neuropsychol. 1992;14:61-975.
[ Links ]DIFIGLIA M. Excitotoxic injury of the neostriatum: A model for Huntington's Disease. Trends Neurosci. 1990; 7:286-289.
[ Links ]DUNNETT SB, CARTER RJ, WATTS C, TORRES EM, MAHAL A, MANGIARINI L, et al. Striatal transplantation in a transgenic mouse model of Huntington'Disease. Exp Neurol. 1998;154:31-40.
[ Links ]DURE LS, YOUNG AB, PENNEY JB. Excitatory amino acid binding sites in the caudate nucleus and frontal cortex of Huntington's Disease. Ann Neurol. 1991;30:785-93.
[ Links ]EMERICH DF, WINN SR, HANTRAYE PM, PESCHANSKI M, CHEN EY, CHU Y, et al. Protective effect of encapsulated cells producing neurotrophic factor CNTF in a Monkey model of Huntington's Disease. Nature. 1997;386:395-399.
[ Links ]EHRNHOEFER DE, BUTLAND SL, POULADI MA, HAYDEN MR. Mouse models of Huntington disease: Variations on a theme. Dis Model Mech. 2009;2:123-129.
[ Links ]FEIGIN A, KIEBURTZ K, SHOULSON I. Treatment of Huntington's disease and other choreic disorders. En: Kurlan MD (Ed). Treatment of movement disorders. Philadelphia: Lippincott Company; 1995. p. 337-364.
[ Links ]FEIGIN A. Advances in Huntington's disease: implications for experimental therapeutics. Curr Opin Neurol. 1998;11:357-362
[ Links ]FERRANTE RJ, KUBILUS JK, LEE J, RYU H, BEEZEN A, ZUNC KER B, et al. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington' disease mice. J Neurosci. 2003;23:9418-9427.
[ Links ]FERRANTE RJ. Mouse models of Huntington's disease and methodological considerations for therapeutic trials. Biochim Biophys Acta. 2009;1792:506-520.
[ Links ]FINK JS, SCHUMACHER JM, ELLIAS SL, PALMER EP, SAINT-HILAIRE M, SHANNON K, et al. Porcine xenografts in Parkinson' Disease and Huntington Disease patients: Preliminary results. Cell Transplant. 2000;9:273-278.
[ Links ]FREEMAN TB, HAUSER RA, SAMBER PR, SAPORTA S. Neural transplantation for the treatment of Huntington's Disease. Prog Brain Res. 2000;127:405-411.
[ Links ]GARCÍA DE YÉBENEZ J, ASTARLOA R, RUÍZ PG, SÁNCHEZ V, MARTÍNEZ FJ, PEREIRO FJ, et al. Test predictivo de enfermedad de Huntington. Datos preliminares. Neurología. 1992;7:345-349.
[ Links ]GERFEN CR. The Neostriatal mosaic: Multiple levels of compartmental organization in the basal ganglia. Annu Rev Neurosci. 1992;15:285-320.
[ Links ]GIFFARD RG, BRUNO VMG, AMAGASU SM, CHOI DW, DUGAN LL. NMDA receptors activation results in hydroxyl radical poduction in primary murino cortical cultures. Soc Neurosci. 1992;18:6461-6466.
[ Links ]GRAYBIEL AM. Neurotransmitters and Neuromodulatorin the Basal Ganglia. Trends Neurosci. 1990;13:244-254.
[ Links ]GOULD DH, GUSTINE DL. Basal ganglia degeneration, myelin alterations, and enzyme inhibition induced in mice by the plant toxin 3-nitropropanoic acid. Neuropathol Appl Neurobiol. 1982;8:377-93.
[ Links ]HARPER PS. The epidemiology of Huntington's disease. En: Harper PS (Ed.) Huntington's disease. New York; 1996. p. 201-239.
[ Links ]HARPER SQ. Progress and challenges in RNA interference therapy for Huntington disease. Arch Neurol. 2009;66:933-938.
[ Links ]HARPER SQ, PATRICK DS, XIAOHUA HE, STEREN LE, INES HM, QINWEN M, et al. RNA interference improves motor and neuropathological abnormalities in Huntington disease mouse model. Proc Natl Acad Sci U S A. 2005;102:5820-5825.
[ Links ]HAMILTON BF, GOULD DH. Nature and distribution of brain lesions in rats intoxicated with 3-nitropropionic acid: A typeofhypoxic (energic deficient) brain damage. Acta Neuropathol. 1987;72:286-297.
[ Links ]HANTRAYE P, RICHIE D, MAZIERE M, ISACSON O. A primate model of Huntington's disease: Behavioral and anatomical studies of unilateral excitotoxic lesions of the caudate-putamen in the baboon. Exp Neurol. 1990;108:91-104.
[ Links ]HENG MY, DETLOFF PJ, ALBIN RL. Rodent genetic models of Huntington disease. Neurobiol Dis. 2008;32:1-9.
[ Links ]HERSCH SM, ROSAS HD. Neuroprotection For Huntington Disease: Ready, Set, Slow. Neurotherapeutics. 2008;5:226-236.
[ Links ]HUNTINGTON G. Recollections of Huntington Chorea as I saw it at East Hampton, Long Island, during my boyhood. J Nerv Ment Dis. 1909;37:255-257.
[ Links ]IMARISIO S, CARMICHAEL J, KOROLCHUK V, CHEN CW, SAIKI S, ROSE C, et al. Huntington's disease: from pathology and genetics to potential therapies. Biochem J. 2008;412:191-209.
[ Links ]KARPUJ MV, BECHER MW, SPRINGER JE, CHABAS D, YOSSEF S, PEDOTTI R et al. Prolonged survival and decreases abnormal movements in transgenic model of huntington disease, with administration of the transglutaminase inhibitor cystamine. Nat Med. 2002;28:143-149.
[ Links ]KEMP JA, FOSTER AC, LEESON PD, PRIESTLEY T, TRIDGETT R, IVERSEN LL. 7-chlorokynurenic acid is a selective antagonist at the glycine modulatory site of the N-methyil-D-aspartate receptor complex. Proc Natl Acad Sci U S A. 1988;85:6547-6550.
[ Links ]KEENE CD, CHANG RC, LEVEREZ JB, KOPYOV O, PERLMAN S, HEVNER RF, et al. A patient with Huntington's disease and long-surviving fetal neural transplant that developed mass lesions. Acta Neuropathol. 2009;117:329-339.
[ Links ]KIEBURTZ K, FEIGIN A, MCDERMOTT M, CARNO P, ABWENDER D, ZIMMERMAN C, et al. A controlled trial of remacemide hydrochloride in Huntington's Disease. Mov Disord. 1996;11:273-277.
[ Links ]KIM M, LEE ST, CHU K, KIM SU. Stem cell-based cell therapy for Huntington disease: A review. Neuropathology. 2008;28:1-9.
[ Links ]KOPYOY OV, JACQUES S, LIEBERMAN A, DUMA CM, EAGLE KS. Safety of intrastriatal neurotrasplantation for Huntington's Disease patients. Exp Neurol. 1998;149: 97-108.
[ Links ]KOWALL NW, FERRANTE FJ, MARTIN JB. Pattern of cell loss in Huntington's disease. Trends Neurosci. 1987;10:24-9.
[ Links ]KUHN R, SCHWENK F, AGUET M, RAJEWSKY K. Inducible gene targeting in mice. Science. 1995;269:1427-1429.
[ Links ]LA FONTAINE MA, GEDDES JW, BANKS A, BUTTERFIELD DA. Effect of exogenous and endogenous antioxidants on 3-Nitropropionic acid-induced in vivo oxidative stress and striatal lesion. J Neurochem. 2000;75:1709-1715.
[ Links ]LAIRD CD. Proposed genetic basis of Huntington's disease. Trends Genet. 1990;6:242-247.
[ Links ]LESCAUDRON L. Autologous adult bone marrow stem cell transplantation in an animal model of Huntington's Disease: Behavioral and morphological outcomes. Int J Neurosci. 2003;113:945-956.
[ Links ]LODGE DG, GOLINGRIDGE G. The pharmacology of excitatory amino acid. Trends Pharmacol Sci. 1990;11:81-86.
[ Links ]MA B, CULVER BP, BAJ B, TONGIORGI E, CHAO MV, TENESE N. Localization of BDNF mRNA with the Huntington's disease protein in rat brain. Mol Neurodegener. 2010;5:22-27.
[ Links ]MADRAZO I, FRANCO-BOURLAND RE, CUEVAS C, CASTREJON H, CUEVAS C, OSTROSKY-SOLIS F. Fetal Neural grafting for the treatment of Huntington'Disease (HD)-report of the first case. Soc Neurosci Abstr. 1991;17:902-907.
[ Links ]MANHOOD A, LU D, YI L, CHEN JL, CHOOP M. Intracranial bone marrow transplantation after traumatic brain injury improving functional outcomes in adult rats. J Neurosurg. 2001;194:589-595.
[ Links ]MARSDEN CD. Trastornos del Movimiento. En: Weatherll DJ, Ledingham JGG, Warrell DA, editores. Oxford tratado de Medicina Interna. Tomo 4. Segunda Edición. Oxford: Editorial Médica Panamericana S A. Ediciones Médicas Folium; 1993. p.3630-3656.
[ Links ]MARTIN B, GOLDEN E, KESELMAN A, STONE M, EGAN JM. Therapeutic perspectives for the treatment of Huntington's disease: Treating the whole body. Histol Histopathol. 2008;23:237-250.
[ Links ]MASON ST, FIBRIGER HC. Kainic acid lesions of the striatum: behavioural sequalae similar to Huntington's chorea. Brain Res. 1978;155:313-329.
[ Links ]MAYER MI, WESTBROOK GL, GUTHRIE PB. Voltage dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature. 1984;309:462-465.
[ Links ]MCDONALD ME, GUSELLA JF. Huntington's disease; translating CAG repeal into a pathogenic mechanism. Curr Opin Neurobiol. 1996;6:638-43.
[ Links ]MEZEY E, CHANDROSS KJ, HARTA G, MAKI RA, MCKERCHER SR. Turning blood into brain: Cells bearing neuronal antigens generated in vivo from bone marrow. Science. 2000;290:1779-1782.
[ Links ]MITCHELL IJ, LAWSON S, MOSER B, LAIDLAW SM, COOPER AJ, WALKINSHAW G, et al. Glutamate-induced apoptosis result in a loss of striatal neurons in the parkinsonian rat. Neuroscience. 1994;63:1-5.
[ Links ]MULLER FJ, SNYDER EY, LORING JF. Gene therapy: can neural stem cell deliver?. Nat Rev Neurosci. 2006;7:75-84.
[ Links ]MURMAN DL, GIORDANI S, MELLOW AM, JOHANNS JR, LITTLE RJ, HARIHAMAN M. Cognitive, behavior and motor effects of the NMDA antagonist ketamine in Huntington's disease. Neurology. 1997;49:153-161.
[ Links ]NAKANISHI S. Molecular diversity of glutamate receptors and implications for brain function. Science. 1992;258:597.
[ Links ]NOVELLI A, REYLLY JA, LYSKO PG, HENNEBERRY RC. Glutamate becomes neurotoxic via the N-methyl-D-aspartate receptor when intracellular energy levels are reduced. Brain Res. 1988;451:205-212.
[ Links ]OLNEY JW, HO OL, RHEE V. Cytotocic effects of acidic and sulphur containg amino acids the infant mouse central nervous system. Exp Brain Res. 1971;14:61-76.
[ Links ]OLIVEIRA JMA. Nature and cause of mitochondrial dysfunction in Huntington's disease: focusing on huntingtin and the striatum. J Neurochem. 2010;114:1-12.
[ Links ]PALFI S, FERRANTE RJ, BROUILLE E, BEAL MF, DOLAN R, GUYOT MC, et al. Chronic 3-Nitropropionic Acid Treatment in Baboons replicates the cognitive and motor deficits of Huntington's Disease. J Neurosci. 1996;16:3019-3025.
[ Links ]PARR AM, TATOR CH, KEATING A. Bone Marrow-derived mesenchymal stromal cells for the repair of central nervous system injury. Bone Marrow Transplant. 2007;40:609-619.
[ Links ]PEREZ-NAVARRO E, CANUDAS AM, AKERUND P, ALBERCH J, ARENAS E. Brain-derived neurotrophic factor, neurotrophin-3, and neurotrophin-4/5 prevent the death of striatal projection neurons in a rodent model of Huntington's disease J Neurochem. 2000;75:2190-2199.
[ Links ]PETERSÉN A, HULT S, KIRIK D. Huntington's disease - new perspectives based on neuroendocrine changes in rodent models. Neurodegener Dis. 2009;6(4):154-164.
[ Links ]PERRY GM, TALLAKSEN-GREENE S, KUMAR A, HENG MY, KNEYNSBERSG A, VAN GROEN T, et al. Mitochondrial calcium uptake capacity as a therapeutic target in the R6/2 mouse model of Huntington's disease. Hum Mol Genet. 2010;19:3354-3371.
[ Links ]PROCKOP DJ. Marrow stromal cell as stem cell for nonhematopoietic tissues. Science. 1997;276:7-4.
[ Links ]QUINN N, BROWN R, CRAUFURD D, GOLDMAN S, HODGES J, KIEBURTZ K, et al. Core Assessment Program for intracerebral transplantation in Huntington's Disease. (CAPIT-HD). Mov Disord. 1996;11:143-50.
[ Links ]QUINTANILLA RA, JOHNSON GV. Role of mitochondrial dysfunction in the pathogenesis of Huntinton' disease. Brain Res Bull. 2009;80:242-247.
[ Links ]REINIER RL, ALBIN HD A, AMATO CJD, PENTNEY JB, YOUNG AB. Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci U S A. 1998;85:5733-5737.
[ Links ]RODRIGUEZ MC, RODRIGUEZ M, GURIDI J, ALVAREZ L, OBESO A. Características Clínicas y fisiopatológicas de la enfermedad de Parkinson. Base anatomofuncional. En: Tratado sobre la enfermedad de Parkinson. Editores: Grandas F, Obeso JA, Tolosa E. Madrid: Editado y coordinado para la 3ra edición LUZAN 5, Ediciones Madrid; 2004. p. 27-38.
[ Links ]RAMASWAMY S, MCBRIDE JL, KORDOWER JH. Animal Models of Huntington's Disease. ILAR J. 2007;48:356-373
[ Links ]SÁNCHEZ-RAMOS J, SONG S, CARDOZO PELAEZ F, HAZZI C, STEDEFORD T, WILLING A, et al. Adult bone marrow stromal cells differentiate into neural cell in vitro. Exp Neurol. 2000;164:247-256.
[ Links ]SCHOEPP DD, CON PJ. Metabotropic glutamate receptors in brain function and pathology. Trends Pharmacol Sci. 1993;14:13-20.
[ Links ]SCORTTICATI MC, MICHELI F. Historia de la enfermedad de Parkinson. En: Micheli EF, editor. Enfermedad de Parkinson y trastornos relacionados. Buenos Aires; 1998. p.1-7.
[ Links ]SCOLDING N, MARKS D, RICE C. Autologous mesenchymal bone marrow stem cells: Practical considerations. J Neurol Sci. 2008;265:111-115.
[ Links ]SPENCER PS, ROY DN, LUDULPH A, HUGON J, DWIVEDI MP, SCHAUMBURG HH. Lathyrism: Evidence for a role of thee neuroexitatory amino acid BOAA. Lancet. 1987;11:1066-1067.
[ Links ]SRAMBA M, RATTAJ M, MOLINAH, VOJTSSAK J, BELAN V, RUZICKY E. Stereotactic Technique and pathophysiological mechanisms of neurotrasplantation in Huntington Chorea. Stereotact Funct Neurosurg. 1992;2:79-83.
[ Links ]STELLER H. Mechanisms and genes of cellular suicide. Science. 1995;267:1445-1449.
[ Links ]STOREY E, BEAL MF. Neurochemical substrates of rigidity and chorea in Huntington's desease. Brain. 1993;116:1201-1222.
[ Links ]SVENDSEN CN, SMITH AG. New Prospects for human stem cell therapy in the nervous system. Trends Neurosci. 1999;22:357-64.
[ Links ]TANAKA M, MACHIDA Y, NIU S, IKEDA T, JANA N, DOI H, et al. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat Med. 2004;10:148-154.
[ Links ]TASSET I, SÁNCHEZ F, TÚNEZ I. Bases moleculares de la enfermedad de Huntington: papel del estrés oxidativo. Rev Neurol. 2009;49:424-429.
[ Links ]THE HUNTIngton's DISEASE COLLABORATIVE RESEARCH GROUP. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes Cell. 1993;72:971-983.
[ Links ]TKAC I, KEENE CD, PFEUFFER J, LOW WC, GRUETTER R. Metabolic changes in quinolinic acid-lesioned rat striatum detected non-invasively by in vivo (1) H NMR spectroscopy. J Neurosci Res. 2001;66:891-898.
[ Links ]TÚNEZ I, SANTAMARÍA A. [Model of Huntington's disease induced with 3nitropropionic acid] Rev Neurol. 2009;48:430-434.
[ Links ]VONSATTEL JPG. Huntington disease models and human neuropathology: similarities and differences. Acta neuropathol. 2008;115:55-69.
[ Links ]WEISS JH, HARTLEY DM, KOH J, CHOI DW. The calcium channel blocker nifedipine attenuates slow exitatory amino acid neurotoxicity. Science. 1990;247:1474-1477.
[ Links ]WONG EHF, KEMP JA, PRIESTLEY T, KNIGTH AR, WOODRUFF GN, IVERSEN LL. The anticonvulsant MK-801 is a potent N-methyl-D-aspartate antagonist. Proc Natl Acad Sci U S A. 1986;83:7104-7108.
[ Links ]WOODBURY D, SCHWARZ EJ, PROCKOP DJ, BLACK IB. Adult rat and human bone marrow stromal cells differentiate into neurons. J Neurosci Res. 2000;61:364-370.
[ Links ]YAMAMOTO A, LUCAS JJ, HEN R. reversal of neuropathology and motor dysfunction in a condicional modelo f Huntington's disease. Cell. 2000;101:57-66.
[ Links ]ZHAO LR, DUAN LR, REYES WM, KEENE M, VERFAILLIE DCM, LOW WC. Human bone marrow stem cells exhibit neural phenotypes and ameliorate neurological deficits after grafting into the ischemic brain of rats. Exp Neurol. 2002;174:11-20.
[ Links ]ZORUMSKI CF, OLNEY JW. Excitotoxic neural damage and neuropsychiatric disorders. Pharmacol Ther. 1993;59:145-162.
[ Links ]
