











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCTION
Cassava is the fourth most important commodity after rice, wheat and corn; a major component in the diet of more than 1 billion people and one of the world's largest source of energy for both human and animal consumption (FAO, 2013). The frogskin disease is one of the main limiting factors for cassava production in Colombia as it directly affects the roots, causing losses in yield up to 90% (Álvarez et al. 2009; Carvajal-Yepes et al. 2014). In addition, it has been reported in Brazil, Costa Rica, Panama, Perú and Venezuela (Oliveira et al. 2014; de Souza et al. 2014; Carvajal-Yepes et al. 2014) and has been associated with both virus (Pineda & Lozano, 1981; Calvert et al. 2008), and phytoplasmas (16SrIII-L) (Álvarez et al. 2003; 2009).
To advance in the knowledge of the disease´s etiology and to understand the effect that a microorganism causes in the plant, it is necessary to achieve its isolation and to develop pathogenicity tests. The main limitations for this study are the phytoplasma isolation and cultivation, for which attempts have been carried out over 40 years ago without success (Hampton et al. 1969; McCoy, 1979; Lee & Davis, 1986).
Some reports of phytoplasma isolation (Fudl-Allah et al. 1972; 1973; Ghosh et al. 1975) failed to demonstrate the identity of the isolated organism. In addition, Skripal & Malinovskaia (1984), working with the CMIMB-72 medium reported more than one hundred phytoplasma isolations from different plants, mainly with “stolbur" disease. Subsequently, Poghosyan and Lebsky (2004), using the same medium, reported the isolation of “stolbur” phytoplasmas in potato and tomato. In 2012, Contaldo et al., were able to grow phytoplasmas of several ribosomal subgroups in a culture medium commercially available from Mycoplasma Experience Ltda., until 2014.
The present research was carried out to achieve the isolation of 16SrIII-L phytoplasmas detected in cassava affected by the frogskin disease.
MATERIALS AND METHODS
Isolation. From 2013 to 2015 phytoplasma isolation trials were carried out from different tissues of cassava affected by the frogskin disease (Álvarez et al. 2009). The roots were washed with water to remove excess of dirt and soil and subsequently cut into fragments of 20 cm after epidermis removal. Then they were cut lengthwise to expose the vascular cylinder of the root (phloem and xylem) and finally, these tissues were cut into 5 mm fragments with a sterile scalpel. In addition, tissues such as cuttings, petioles, stems and leaves were processed in similar manner, and washed with sterile distilled water.
In the laminar flow chamber, tissue fragments were wetted with 0.5mL of PivL® (Phytoplasma in vitro liquid medium) with phenol red for two minutes. Then the tissues were transferred to a Vacuette tube (Greiner Bio-One, ref. 45001, K) with 4mL of PivL and incubated at 30ºC ±1ºC. In vitro cassava shoots obtained from micropropagation were used as negative control, as was the uninoculated medium.
Tubes were evaluated until observing the change of color, turning from orange - red (pH 7) to yellow (pH below 6.8), indicative of microorganism growth. From each of the tubes that changed color, 150μL were removed and transferred to 6cm diameter Petri dishes, which contained 8mL of PivS® (Phytoplasma in vitro solid medium) and gently dispersed. The Petri dishes were then incubated in an atmosphere of 5% CO2 and 95% N2 for 5 to 7 days 25°C ± 1°C in an anaerobiosis jar containing one AnaeroGen™ envelope which absorbs oxygen and an anaerobiosis indicator (Oxoid Ltd., Basingstoke, Hampshire, UK).
Additionally, phytoplasma isolation was carried out also from periwinkle [Catharanthus roseus L (G Don)] midrib tissue, with symptoms of leaf yellowing, stunting, shoot proliferation, flower malformation.
Molecular identification of phytoplasmas. To detect and identify phytoplasma presence and growth in the liquid medium, from each color-changing tube, 1mL aliquots were transferred into eppendorf tubes and centrifuged at 16,000 rpm for 30 minutes in Thermo Scientific Heraeus Pico 17 MicroCentrifuge, 24-Pl Rotor; 120V. The supernatant was discarded, the precipitate was suspended in 20μL of water and aliquots of 2μL were used for PCR and qPCR tests. Colonies grown in PivS® medium were individually removed with 10μL tips. Then directly employed for PCR and qPCR assays.
In the PCR tests, a mixture of 12.5μL of Go taq master mix 2X Promega (Madison USA), 0.5μL of each primer (final concentration 0.2μM), 9.5μL of water and 2μL of liquid medium or an individual colony transferred from the medium, as reported above were used.
The direct PCR reaction was carried out using the universal P1/Tint primers (Deng & Hiruki, 1991; Smart et al. 1996), which amplify a fragment of about 1,600 bp located in the 16S rDNA region, spacer region, and part of the 23S rDNA. For the nested PCR primers R16F2n/R16R2 (Gundersen et al. 1996; Lee et al. 1993), which generate amplicons of about 1.200 bp corresponding to part of the 16Sr region internal to the previous one were used. The amplification profile was initial denaturation at 94°C for 1 minute and 35 denaturation cycles at 94°C for 1 min, annealing at 50°C for 2 min and extension at 72°C for 3 min, followed by a final extension at the same temperature for 10 min. For the nested PCR, 1μL of the amplified product was diluted in 29μL of water and 2μL were employed for amplification under the same conditions as the direct PCR.
A nested PCR specific for phytoplasmas in 16SrIII group was also performed with R16(III)F2/R16(III)R1 primers (Lee et al. 1994) amplifying a 800 bp fragment in the 16Sr gene. For this amplification, the first denaturation was done during 2 min at 94°C but all the other parameter were as described above.
In the case of the cultures from periwinkle tissues, a nested PCR was performed with universal primers 16R738/16R1232 amplifying a fragment of approximately 500 bp in the 16Sr RNA gene (Gibb et al. 1995).
Seven μL of PCR products were separated on a 1.2% of agarose gel, stained with 4μL/200cc SYBR safe (Invitrogen) and visualized under transilluminator (Sambrook et al. 1989). Subsequently, the amplicons were cleaned using the DNA precipitation method with polyethylene glycol and ethanol (Ziv & Fuentes, 2007), and sequenced at the Iowa State University using primers R16(III)F2/R16(III)R1 and 16R738/16R1232 for cassava and periwinkle isolates, respectively.
Sequences were assembled with the Chromas Pro 1.5 program, compared by maximal mating and homology higher percentages to those of selected phytoplasma strains in GenBank, using the BLAST tool of the National Biotechnology Information Center (Altschul et al. 1990). They were also aligned with the Clustal 6.0 program, and deposited at the GenBank.
In addition, qPCR assays were made for the detection of 16SrIII phytoplasmas, following the methodology developed in the Plant Pathology Lab (CIAT). The probe and primers are based on the 16S rRNA gene within a 1,400 bp region: in particular, the probe is at position 469 and the primers at 416 (sense) and 492 (antisense), achieving the amplification of a fragment of 72 bp and both used at 200nM.
Microscopy. The colonies were observed directly into the Petri dish under light binocular microscopy (Leica DM500 and camera Leica ICC50 HD) at magnifications from 10X to 100X.
For the electron microscopy observation, the colonies were washed with sterile distilled water and 1mL was deposited into sterile Eppendorf tubes; cell precipitation was obtained by centrifugation at 16,000 rpm for 30 minutes. After discarding the supernatant, the samples were fixed in 2.5% glutaraldehyde and sent to the Santafé clinic (Bogotá, Colombia) for further processing. Post-fixation was done in 1% osmium tetraoxide, with centrifugations at 13,000 rpm for 3 minutes at each step. Subsequently dehydratation in an ethanol upward gradient from 50 to 100%, with ethanol-acetone mixtures was performed before the final embedding in epoxy resin (Sigma-Germany). The polymerization was performed at 70°C for 48 hours. Ultrathin sections between 60 and 150nm were obtained with (Leica Ultracut - Germany) ultramicrotome, they were contrasted with uranyl acetate and lead citrate and observed under Jeol 1400 plus transmission electron microscope at 120 kV.
Complementary PCR tests. To confirm that the identity of the microorganism grown in the medium was a phytoplasma and not contaminating microorganisms, specific PCR tests were performed for ‘Candidatus Liberibacter’ spp., Rickettsias and universal bacteria, because these microorganisms have a great morphological similarity with phytoplasm. For ‘Ca. Liberibacter’ the PCR reaction was made with specific OI2c-OI1 primers, which amplify a fragment of 16S rDNA of 1,160 bp (Jagoueix et al. 1994). As positive control, a citrus DNA sample was used, provided by Saulo Alves Santos de Oliveira (Cassava and Fruticulture, Brazilian Agricultural Research Corporation; EMBRAPA, Brazil). The amplification thermal profile was as reported by Jagoueix et al. (1996).
In the case of rickettsia, the samples were amplified using primers CS-78 and CS-323, which amplify a 401bp of the citrate synthase gene (gltA), reported for the detection of Rickettsia spp., by Labruna et al. (2004). Amplification conditions were initial denaturation of three min at 95°C, followed by 40 cycles of denaturation at 95°C for 15 sec, annealing at 48°C for 30 sec, extension at 72°C for 30 sec and a final extension of seven minutes at 72°C. The positive control consisted of a DNA sample of Rickettsia sp. (strain: Atlantic rainforest), kindly provided by Juan David Rodas G., University of Antioquia, Medellín, Colombia.
Bacterial detection was done with universal primers amplifying a 16Sr fragment of 1,600 bp EubB27/EubA1522. The thermic profile was denaturation at 96°C for 3 min followed by 35 cycles of denaturation at 96°C for 1 min, annealing at 55°C for 1 min, extension at 72°C for 3 min and a final extension at 72°C for 7 min (Giovannoni, 1991; Suzuki & Giovannoni, 1996).
RESULTS AND DISCUSSION
Growth in liquid medium. The phytoplasma isolation in PivL® culture medium allowed the growth of cassava frogskin phytoplasmas and pigeon pea witches' broom-related phytoplasmas, from cassava and periwinkle tissues respectively. Experiments showed positive results when tissue from root phloem (Figure 1), cassava cuttings and, periwinkle stems were used. The tubes changed color after 5 to 15 days. No color change was recorded in the other tissues, micropropagated shoots and the medium without inoculation. The nested PCR confirmed the phytoplasma presence by amplifying expected lenght fragments with primers R16 (III) F2/R16 (III) R1 from cassava and universal primers 16R738/16R1232 from periwinkle inoculated liquid media. In addition, the positive results of qPCR test with probe and specific primers for cassava frogskin phytoplasma further confirmed the phytoplasmas presence in the samples processed from liquid medium with color change (Figure 2).

Figure 1 a. Cassava root with frogskin disease, note the brown shade of the vascular tissue where the phloem beams are concentrated; b. Left tube, root fragments in PivL® medium eight days after sowing and right the negative control containing uninoculated medium; c. Left tube, negative control and right tube, root fragments of healt plant in PivL®.

Figure 2 Real-time PCR of samples of PivL medium seeded with cassava root tissue. Real-time PCR plot with probe and specific primers for the 16SrIII group, indicating that eight of the nine liquid medium samples (PivL) with cassava root tissue were positive for phytoplasma. The positive control corresponded to a plasmid of Cassava frogskin disease phytoplasma.
Contaldo et al. (2016), showed that is possible phytoplasma isolation from infected plants-host field-collected. In this investigation, the times recorded for the color change in the PivL® medium were much shorter than those achieved in the study of Contaldo et al. (2012), ranging from 16 to 90 days. However, the same authors propose that the difference in time elapsed for the color change may be due to the type of phytoplasma that is being isolated or its concentration in the tissues.
In these experiments conducted with cassava, the liquid medium changed color consistently when plant tissue used was from frogskin diseased materials, but never with healthy roots, leaves, petioles or stems and this is a further evidence of the phytoplasma association with the disease.
Growth in solid medium. Microscopic examination of Petri dishes with PivS® medium from both cassava and periwinkle tissues showed the presence of colonies similar to those formed by mycoplasmas and to those already reported for phytoplasmas (Figures 3 y 4). Colonies are translucent, rounded and difficult to observe with the naked eye. These appear from the second day of incubation and after a week or more, they increase in number. The colonies obtained from cassava and periwinkle tissues are similar in morphology and size. Some colonies present the typical morphology of fried egg, but are mostly round colonies, smooth, convex and towards the end of the colony form a double border of lighter color.

Figure 3 Colonies grown in PivS medium from PivL, in an atmosphere of 95% N2 and 5% CO2, photographed between 40 and 100X. A, B and C: colonies obtained from cassava tissues; D and E: colonies obtained from periwinkle tissues. A) Colonies at two day incubation; B) Single colony at 5 days of incubation; C) Individual colony at 12 days of incubation; D) Colonies at 3 days incubation; E) Colonies at 8 days incubation.

Figure 4 Colony of Phytoplasma grown in PivS, obtained from cassava root tissues. Photographed a 100X.
The colonies obtained are similar to those reported for mycoplasmas (Tully, 1993; 1995; Pitcher et al. 2005) and for phytoplasmas (Poghosyan & Lebsky, 2004; Contaldo et al. 2012; 2016).
Molecular analyses on individual colonies grown in solid medium were positive after R16F2n/R2 PCR tests, followed by nested PCR with universal primers 16R738/16R1232 in the case of periwinkle, and R16 (III) F2/R16 (III) R1 in the case of cassava.
The phytoplasma presence was also confirmed by the qPCR tests with specific primers for phytoplasmas of 16SrIII group, which allowed to verify the presence of cassava frogskin phytoplasma in these colonies.
Sequence analyses of amplified fragments with specific R16 (III) F2/R16 (III) R1 and universal 16R738/16R1232 primers showed 99 to 100% homology with phytoplasmas reported in the GeneBank from cassava frog skin phytopalasma and periwinkle with group 16SrIX phytoplasmas respectively. In the case of cassava isolates, sequences of 800bp were obtained from liquid medium and individual colonies in solid medium and four of them were deposited in the GeneBank under (accession numbers: KT780862 - KT780865). Also, liquid and solid media inoculated from symptomatic periwinkle showed 99% homology with pigeon pea witches' broom phytoplasma (16SrIX group) (data not shown).
Results obtained in this study are in agreement with those reported by Contaldo et al. (2012; 2016), demonstrating that phytoplasmas growth can be achieved also from other phytoplasmas from field infected materials such as those belonging to groups 16SrIII and 16SrIX. This metodology will serve in the future as a complement to the diagnosis of plant infections associated with these microorganisms and may be essential for their biological, biochemical and molecular characterization, including pathogenicity and genetic resistance testing by eperimental inoculation.
In agreement with recent results (Contaldo et al. 2016) the incubation of the solid medium in an atmosphere saturated with nitrogen and carbon dioxide result to be essential to the obtaining of colonies, since when it was not used, there were contamination problems by other bacteria. The explanation possibly lies in the anaerobic growth condition of phytoplasmas and the use of nitrogen in the electron transport chain to the cell membrane.
The complex reductive evolution of Mollicutes, has led them to the loss of genes for amino acids and cell wall synthesis, among others, these losses require the use of complex culture media for their isolation, which must contain sterols, fatty acids, nucleotides, amino acids and other compounds that meet their nutritional and osmotic pressure requirements (Tully, 1995; Jensen et al. 1996; Meseguer et al. 2012). Analogously, the culture media described in phytoplasma isolation studies contain the same and other elements (Fudl Allah et al. 1972; 1973; Ghosh et al. 1975; Skripal & Malinovskaia, 1984). On the other hand, the recently reported medium CB (Contaldo et al. 2016) is quite similar to the medium employed for mycoplasma growth furhter confirming the close relationship between phytoplasmas and some of the human infecting mycoplasmas (Gasparich et al. 2004).
Currently there are a total sequencing of five ‘Candidatus Phytoplasmas’ (‘Ca. P. asteris' strains OY-M and AY-WB, two strains of 'Ca. P. australiense' and one of 'Ca. P. mali') indicating that their genome size is below 900 b (Kube et al. 2012). This makes the isolation of phytoplasmas even more difficult, since, in addition to the absence of many genes involved in the amino acids synthesis, nucleotides, fatty acids, tricarboxylic acid cycle and oxidative phosphorylation, there is the lack of genes needed for the pentose phosphate pathway and subunits of ATP synthase, essential for life (Oshima et al. 2004). This could also explain why there are more successful cases of isolation of mycoplasmas than isolation of phytoplasmas.
Electron Microscopy. The electron microscopy observations of embedded materials allowed to confirm the prokaryotic cells presence with shape and size similar to those observed for phytoplasmas (Figure 5). The predominant morphology was pleomorphic, circular and oval, which is typical of phytoplasmas, with no notable differences between the isolates from cassava and periwinkle. The average size was approximately 500nm in diameter in the rounded cells, and up to 2μm for the pleomorphic ones (data not shown).
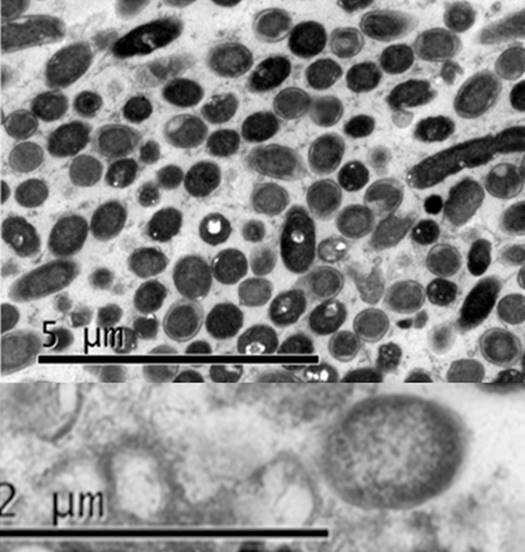
The electron microscopy observations showed in colonies the presence of prokaryotic cells with shape and size similar to those observed for phytoplasmas (Chapman et al. 2001; Siddique et al. 2001; Pribylova et al. 2001; Musetti et al. 2002; Bertaccini & Lee, 2018) in plant infected tissues. In electron microscopy studies dimensions ranging from 120nm to 500nm in diameter and 1.2μm or longer in length were reported (Bertaccini & Marani, 1980; Waters & Hunt, 1980; Lee et al. 2000). Chapman et al. (2001) claim that the morphological changes in phytoplasma cells heavily depends on the species involved, age, phloem anatomical aspects and other factors.
Complementary molecular tests. The PCR tests with universal primers for ‘Ca. Liberibacter’ spp. and Rickettsia were negative in all cases of individual colonies grown in the PivS culture medium tested, but the positive controls were amplified (data not shown). In the same way, PCR tests with universal primers for other bacterial genera, did not allow the amplification of the expected DNA fragment, indicating that no contamination was present in the colonies. However, in tests performed in the absence of nitrogen and conditions that did not guarantee anaerobiosis, amplification was obtained and after sequencing, bacterial contamination was detected with bacteria identified as enterobacteriaceae (data not shown).
On the other hand, the ‘Ca. Liberibacter’ spp are limited to phloem and Rickettsia is limited to xylem, these pathogens have a great morphological similarity with the cells observed in this study and this was the main reason for carrying out the molecular tests for their detection. Results show that none of these microorganisms is present in the colonies obtained.
In conclusion, the evidence of phytoplasma growth generated in this research was achieved by different techniques such as light and electron microscopy, PCR, qPCR, and sequencing studies, taking into consideration the colonies morphology, size, shape and cellular characteristics, the 16S rRNA gene sequences.
This study allowed to demonstrate the consistent presence of phytoplasma 16SrIII to the phloem of cassava root with frogskin disease. This behavior can be explained by the distribution of phytoplasmas in the plant, being these pathogens forced to restrict themselves to the phloem by the lack of cell wall (Doi et al. 1967; Gundersen et al. 1996; Marcone, 2013), they are reported in many cases as located in the roots, in which sugar sources are mostly accumulated.

