











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
Introduction
Pseudohypoparathyroidism (PHP) is a rare metabolic disease caused by the mutation of the GNAS gene, which encodes the alpha subunit of the stimulating heterotrimeric G protein. This heterozygous mutation is maternally inherited and causes parathormone (PTH) resistance secondary to parathormone receptor type 1 (PTH1R) autoantibodies, and thus inadequate activation of 25-Hydroxy vitamin D through the enzyme 1- alpha-hydroxylase. In addition, PTH resistance results in increased renal reabsorption of phosphorus. All of these factors cause hypocalcemia, hyperphosphatemia, and increased PTH levels in these patients.1
PHP is a rare disease for which there are few studies; consequently, data on this condition are scant; however, Underbjerg et al.2 identified 60 cases in a study attempting to identify all PHP patients in Denmark, representing a prevalence of 1.1 cases per 100 000 population.
Originally, all persons with phenotypic features typical of Albright's hereditary osteodystrophy (rounded facies, short stature, obesity, subcutaneous ossification, shortened fourth metacarpal, etc.) had biochemical alterations such as hypocalcemia, hyperphos-phatemia, and increased PTH levels (typical of PHP). However, it is now known that there are five subtypes of PHP (1a, 1b, 1c, 2, and pseudohypoparathyroidism) and that patients do not have these phenotypic characteristics in two of them (1b and 2).3
Gitelman syndrome (GS) is an autosomal recessive salt-losing renal tubulopathy characterized by metabolic alkalosis with hypokalemia, hypomagnesemia, and hypocalciuria, with an estimated prevalence of 1 case per 40 000 inhabitants.4
The following is the case of a middle-aged man with tetanic seizures since childhood, who met the biochemical criteria for the diagnosis of PHP, without associated phenotypic characteristics. He presented with metabolic alkalosis with hypokalemia and hypocalciuria, as well as renal potassium losses, which are consistent with GS.
This is the second case reported worldwide of a patient who presented concomitantly with these two diseases, both of a genetic nature and with a very low prevalence among the general population.5 Nonetheless, it should be noted that both conditions were diagnosed based on clinical findings, and no genetic confirmation was obtained due to the lack of genetic studies in the country.
Case report
A 37-year-old male migrant with a history of tetanic seizures since the age of 9 years and an unclear diagnosis of hypokalemic periodic paralysis since 2013 (under intermittent treatment with potassium gluconate) was admitted to the emergency department of a secondary care hospital in Bogotá D.C., Colombia, due to a sensation of paresthesia in hands and feet during the last 2 days, which progressed to intermittent painful spasms in skeletal muscles and intermittent muscle contractions.
The patient reported no recent travel, no contact with sick people, no respiratory, gastrointestinal, or cardiovascular symptoms, and no family history of any type of genetic syndrome or altered phosphorus-calcium balance (siblings, parents, and grandparents).
On admission examination, the patient was found in good general condition and with the following vital signs: blood pressure: 110/75 mmHg, heart rate: 98 bpm, respiratory rate: 18 rpm, temperature: 36.3°C, and oxygen saturation: 94%. No abnormal findings were reported on physical examination, but signs of Trousseau and Chvostek were evident. His facies and skeletal features were normal, and he had no thyromegaly or thyroid nodules. The patient weighed 72kg and was 1.75m tall.
Upon admission to the emergency department, laboratory studies were performed to rule out infectious, metabolic, toxic, and electrolyte disorders. The presence of hypocalcemia, hypomagnesemia, hyperphosphatemia, and hypokalemia was notable among the findings of these studies (Table 1). Taking into account what was reported in the emergency room, the patient was hospitalized. On the third day of hospital stay, follow-up tests were carried out, showing elevated PTH levels (369 pg/mL). Consequently, after ruling out several causes of secondary hyperparathyroidism, such as vitamin D deficiency, chronic kidney disease, malabsorption, or chronic liver disease, PTH-dependent hypocalcemia, consistent with PHP, was suspected.
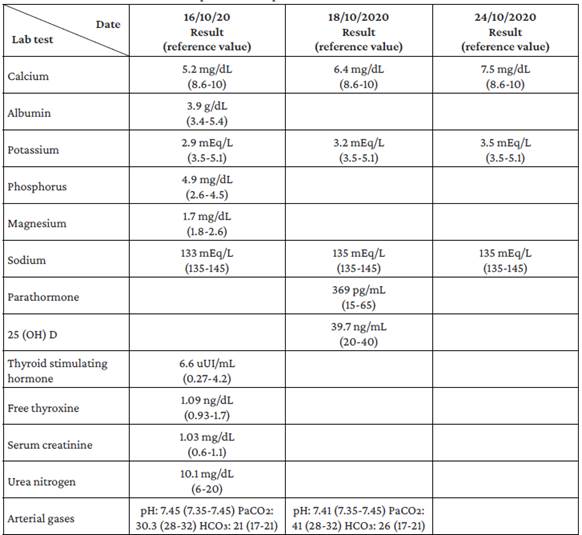
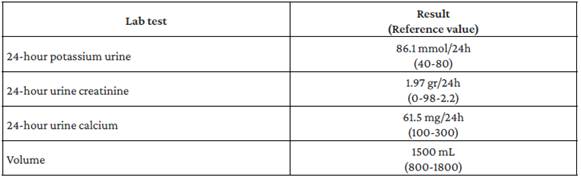
It was determined that the patient had PHP type 1b or 2 due to the absence of dysmorphic facies that would raise suspicion of Albright's hereditary osteodystrophy. However, it was impossible to determine the PHP type because a test to measure nephrogenic cyclic adenosine monophosphate (cAMP) levels could not be performed in the hospital. Simultaneously, the patient presented with metabolic alkalosis, increased renal potassium losses, hypocalciuria, and normal blood pressure (Table 2), so GS was suspected, although it could not be confirmed because the necessary genetic tests were not available at the hospital where he was treated.
From the first day of hospitalization, the patient required intravenous calcium gluconate (1g every 8 hours), potassium chloride (4 mEq per hour), and magnesium sulfate (2g every 12 hours), but on the fifth day, intravenous calcium and potassium replacement were switched to oral supplement (1.2g of calcium carbonate every 8 hours, 0.5 micrograms of calcitriol every 12 hours, and 10mEq of potassium gluconate every 12 hours), thus achieving a satisfactory progress.
The patient was discharged from the hospital after 8 days with resolution of symptoms and stable serum electrolyte levels, and was prescribed outpatient follow-up, electrolyte monitoring, and indefinite oral supplementation, which he began during his hospitalization. The patient returned to the hospital 10 days later for an outpatient follow-up appointment, where he reported that he was still asymptomatic; however, he did not provide the results of the laboratory tests requested at discharge.
Discussion
PHP is a disease characterized by PTH resistance, which manifests biochemically as hypocalcemia, hyperphosphatemia, and elevated PTH levels.1 Although it is a rare disorder, several studies have attempted to establish its prevalence; for example, Linglart et al.2 in a study conducted in Denmark, found a prevalence of 1.1 cases per 100 000 inhabitants and Lopes et at.,6 in a study conducted in Parana, Brazil, in 55 patients with hypoparathy-roidism, reported a prevalence of 9.1%.
When PTH binds to its PTH1R receptor, dissociation of the alpha, beta, and gamma subunits of the stimulating heterotrimeric G protein occurs, with the subsequent activation of adenylyl cyclase and synthesis of cAMP, which activate protein kinase A and then a series of enzymatic cascades that affect various cell functions.7 The GNAS gene encodes the alpha subunit of PTH1R, and inactivating mutations have been described as a genetic and epigenetic cause of PHP.8-10
Historically, five PHP subtypes have been identified, with 1A being the first to be characterized given its association with Albright's hereditary osteodystrophy. Subtypes 1B and 2 have the biochemical characteristics mentioned above, without any phenotypic peculiarity, but are differentiated by nephrogenic cAMP levels after stimulation with PTH.11
In the case presented here, despite not being capable of ruling out an abnormality in PTH structure, the most accurate clinical and biochemical diagnosis is consistent with PHP subtype 1B or 2. Unfortunately, the measurement of nephrogenic cAMP is not available in Colombia and, therefore, it was not possible to determine the specific subtype in this patient; however, it is important to note that this situation prevents the classification of the subtype but not the diagnosis of PHP. This had already been reported by Trejo et al.12 in a case series of 4 patients diagnosed with PHP in adulthood who had hypocalcemia, hyperphosphatemia, elevated PTH levels, and normal renal function. Also, in view of the absence of a family history, a sporadic, non-hereditary mutation was suspected.
Furthermore, the patient had metabolic alkalosis with hypokalemia, accompanied by mild hypomagnesemia, increased renal potassium losses, and hypocalciuria, which is consistent with the diagnosis of GS, a disease in which sodium reabsorption in the distal convoluted tubule is altered due to a mutation of the sodium-chloride cotransporter, resulting in secondary hyperaldosteronism.13 Symptoms in patients with GS usually begin in adolescence and are characterized by metabolic alkalosis with hypokalemia and normal blood pressure, despite hyperaldosteronism, as evidenced in the reported case. Moreover, patients typically present with hypocalciuria and hypomagnesemia, in contrast to Bartter syndrome, in which hypercalciuria is the distinctive feature.14
Hypocalciuria, in cases of GS, is explained by increased calcium reabsorption in the proximal tubule in response to volume depletion caused by sodium loss,15 which was also evident in the reported patient. It should be pointed out, however, that genetic confirmation of this tubulopathy was not possible.
The present report is of great relevance because it describes the case of a patient who presented with two clinical entities of very low prevalence, both concomitantly and independently, and that also have no known genetic relationship, because while in PHP the GNAS mutation is located on chromosome 20,4 in SG the mutated gene is SLC12A3, located on chromosome 16.7 Additionally, according to the literature reviewed for the preparation of this report, only one case of the same patient with these two conditions has been published,5 making this case the second reported case worldwide.
Conclusion
Although it was not possible to obtain genetic confirmation of the diagnoses of PHP and GS in the present case, this may be the second case reported in the world of a patient with clinical suspicion of these two entities. This raises the question of whether this is merely a random and simultaneous occurrence of two conditions of genetic etiology with a very low frequency, or if, on the contrary, the simultaneous occurrence of these conditions may be due to an unidentified genetic mutation.

