











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
INTRODUCCIÓN
La hipercolesterolemia familiar (HF) es un síndrome genético de carácter autosómico dominante ocasionado por mutación en el cromosoma 19. Representa la principal causa genética de enfermedades cardiovasculares prematuras 1. Se estima que el número de pacientes que presentan HF es de 14 a 34 millones de personas, de los cuales el 10% es diagnosticado y únicamente el 5 % es tratado adecuadamente 2.
La enfermedad describen dos fenotipos diferentes. Por un lado, la hipercolesterolemia familiar heterocigota (HFHe), que se caracteriza por la presencia de mutación en un solo alelo, siendo esta la presentación más frecuente 3. Por otro lado, en la hipercolesterolemia familiar homocigota (HFHo) el individuo presenta mutación para ambos alelos, considerándose una alteración codominante autosómica de presentación rara y con mayor riesgo de compromiso cardiovascular en la niñez 4.
El desarrollo de la HF es causado por la alteración en el metabolismo de lípidos, especialmente en el colesterol de baja densidad (LDL). Las mutaciones patogénicas se relacionan con los genes: el receptor de LDL (LDLr), la apolipoproteina B-100 (Apo- B100) y la proteína convertasa subtilisina / kexina tipo 9 (PCSK9), lo que produce la elevación del colesterol y alteración de la vía del receptor LDL en el 80 % de los casos diagnosticados de HFHe 5.
El objetivo de este trabajo es la presentación de una serie de casos de 5 pacientes pertenecientes de un municipio de Quindío (Colombia) con las siguientes coordenadas 4°32'20"N 75°40'21"O, adultos jóvenes y uno en edad pediátrica, quienes fueron diagnosticados con hipercolesterolemia familiar mediante la detección de una mutación del gen codificante para LDLR y LDLRAP1. Al igual que generar una correlación entre la revisión en bases de literatura y serie de casos de pacientes con respecto HF, los cambios en la cotidianidad del paciente y su familia.
MATERIALES Y MÉTODOS
Declaración ética, consentimiento y permisos
Este estudio se realizó bajo los lineamientos de la Declaración de Helsinki y la Resolución 8430 de 1993 y 2378 de 2008 del Ministerio de Salud y Protección Social de Colombia, por las cuales se adoptan las buenas prácticas clínicas. Se brindó la información necesaria, se aclararon dudas.
Se realizó una búsqueda de la literatura usando como términos "hipercolesterolemia familiar genético" en las bases de datos electrónicas PUBMED, Science Direct y SCOPUS. Todos los tipos de diseño de estudio fueron considerados, priorizando los redactados en inglés o español y utilizando nuestras palabras clave.
Los pacientes fueron identificados a partir de la asistencia de la paciente 4 (caso índice) a la consulta del servicio de cardiología del Centro Cardiovascular y Diabetes Más Salud (Armenia, Quindío) a los que se les practicó un examen físico con la elaboración de la respectiva historia clínica para la posterior construcción del árbol genealógico. También se tomaron medidas antropométricas como índice de masa corporal (IMC), muestras de sangre periférica para exámenes paraclínicos como perfil lipídico (HDL, LDL, TGC y CT), función hepática (fosfatasa alcalina, bilirrubina, transaminasas oxalacetica y pirúvica) y análisis genético mediante técnicas de secuenciación de los genes LDLR y LDLRAP1, siendo clasificadas las variantes encontradas de acuerdo con los criterios del American College of Medical Genetics and Genomics (ACMG), al igual que imágenes diagnósticas como rayos X de tórax y pruebas de función cardiovascular que incluyeron ecocardiograma y SPECT cardíaco.
Reporte del caso
Presentamos el caso de cuatro pacientes de la misma familia, quienes presentan pruebas genéticas positivas para hipercolesterolemia familiar de tipo genético.
Paciente 1
Paciente masculino de 10 años, quien fue diagnosticado con hipercolesterolemia familiar homocigota desde la infancia, con pruebas genéticas positivas (tabla 1). Ha recibido tratamiento farmacológico con atorvastatina 10 mg, ezetimibe 10 mg y omega 3. Paciente sin alteraciones cardiovasculares definido por paraclínicos de extensión.

Diagnóstico genético
Paciente 2
Paciente femenina de 39 años, con antecedente de hipercolesterolemia familiar diagnosticada en las adolescencia y pruebas genéticas positivas (tabla 2), con antecedente familiar de enfermedad coronaria (madre), que recibe tratamiento farmacológico con rosuvastatina/ezetimiba 10g / 10 mg, Aspirina 100 mg/día y Carvedilol 6,25 mg cada 12 horas, con poca adherencia a terapia. A la anamnesis se evidencia presencia de disnea mMRC grado 2 asociado a dolor precordial; se realizó ecocardiograma con estrés (Dobutamina), que evidenció isquemia miocárdica en pared infero-posterior y ecografía abdominal con foco de colesterolosis vesicular. Resto de paraclínicos sin alteraciones.

Paciente 3
Paciente masculino de 33 años con antecedente familiar de hipercolesterolemia. Sin afectación orgánica ni tratamiento farmacológico.
En ecocardiograma transtorácico se evidencia: compromiso de movilidad segmentaria del ventrículo izquierdo, con función sistólica disminuida, fracción de eyección del ventrículo izquierdo del 45 % e insuficiencia tricuspídea leve con función diastólica conservada.
En estudio Holter ritmo de 24 horas presenta extrasístoles ventriculares 36 en 24 horas y extrasís-toles supraventriculares 6 en 24 horas, sin fenómenos repetitivos.
El paciente no presenta evidencia de estudios genéticos, sin embargo, refiere son positivos para mutación.
Paciente 4
Paciente femenina de 53 años, con antecedente de obesidad grado I (IMC: 30 kg/m2), hipertensión arterial sistémica, enfermedad coronaria y dislipidemia (tabla 3). En manejo farmacológico con Carvedilol (6.25mg cada 12 horas) y ácido acetilsalicílico (100 mg al día).
Tabla 3 Resumen antecedentes de paciente 4

AMI: Arteria mamaria interna. ADA: Arteria descendente anterior. S: Safena. CX: Circunfleja. RI: Ramo intermedio ECV: Enfermedad cerebro vascular.
Al examen físico se evidencia: obesidad (IMC: 30 kg/m2), xantelasmas palpebrales bilaterales, arco corneal bilateral y exploración del cuello con soplo carotídeo izquierdo. Abdomen con angioqueratomas periumbilicales y extremidades con múltiples xantomas (tabla 4).


Perfil lipídico: CT/LDL: 579/496, TGC/HDL: 187/45.6.
Función hepática: Fosfatasa alcalina: 70.1, bilirrubina total: 0.66, bilirrubina directa: 0.10, bilirrubina total: 0.56. TGO/TGP: 19/15
Perfil auto inmunológico: negativo.
Imágenes
Estudio SPECT cardíaco: Paciente con mala tolerancia al ejercicio a quien se realiza prueba de estrés con dipiridamol, no se evidencian alteraciones isquémicas del segmento ST. Sin embargo, el paciente se queja de dolor precordial opresivo, no arritmias, respuesta presora adecuada. Se concluye que la prueba de estrés con dipiridamol es sospechosa para isquemia miocárdica. La función sistólica del ventrículo izquierdo en el estrés farmacológico y en el reposo está levemente disminuido (FEVI: 48 %).
Ecocardiograma: función sistólica preservada, fracción de eyección (FE): 66 %, volumen de final de diástole (VFD) de 86cc sin trastorno de la relajación. Insuficiencia valvular mitral grado I/IV HTP leve, insuficiencia valvular tricuspídea, resto de examen dentro de parámetros normales.
Radiografía de tórax: dentro de parámetros normales.
Se realizó un cálculo de los criterios de Holanda para el diagnóstico clínico de hipercolesterolemia familiar. Su interpretación fue: diagnóstico de HF: certeza: > 8 puntos; probable: 6-7 puntos; posible: 3-5 puntos. La paciente obtuvo un puntaje de 19 puntos (tabla 5), por lo cual se consideró la realización de un estudio genético para hipercolesterolemia familiar.
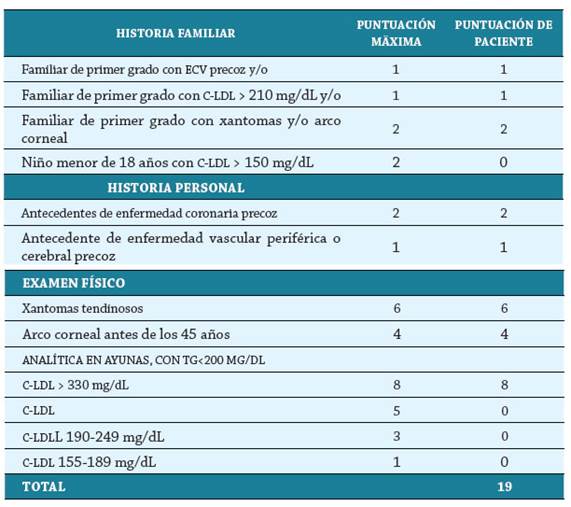
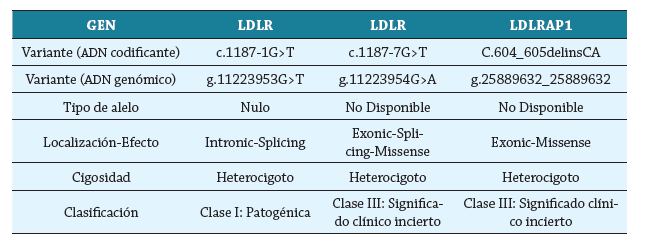
Tabla 6 Caracterización genética del paciente. Se confirma diagnóstico de hipercolesterolemia familiar de carácter heterocigoto secundario a una mutación del gen LDL (patogénico) y una mutación del gen LDLRAP1 (carácter incierto)
Adicionalmente, el grupo familiar recibe abordaje y asesoramiento genético. Se encontró que varios integrantes de la familia tienen alta probabilidad de presentar hipercolesterolemia familiar (figura 2), ya que varios de ellos presentan factores de riesgo y elementos predisponentes, asociado a clínica sugestiva y perfil lipídico confirmatorio.
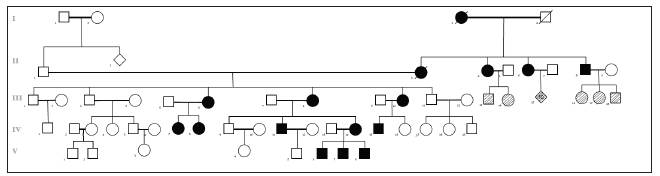
Figura 2 Genealogía del grupo familiar, siendo la paciente III.8 el caso índice (flecha). Se evidencia carácter dominante de la enfermedad por el número de individuos afectados y patrón de herencia. Las figuras en negro representan los pacientes con diagnóstico confirmado por prueba molecular o muy alta sospecha por niveles de LDL en sangre. Las figuras con patrón de cebra representan pacientes posiblemente afectados, sin embargo, no existen datos confirmatorios
DISCUSIÓN
El objetivo de este trabajo es la presentación del caso y generar una correlación clínica y con base en la literatura, con respecto a la HF, evaluar la epidemiología, así como otros aspectos: sospecha clínica, diagnóstico y cotidianidad de los pacientes con diagnóstico genético y los cambios en su estilo de vida, así como de su familia.
El reporte del caso presentado se fundamenta en la sospecha clínica de hipercolesterolemia familiar según los Criterios Holanda en el paciente y posterior confirmación genética o diagnóstico por alta probabilidad debido a niveles séricos de LDL.
Con base en la búsqueda de la literatura se evidencia que en Colombia la mutación encontrada en la serie de casos familiar no ha sido ampliamente estudiada en el país, y por tanto no existe información suficiente para reconocer su incidencia. Puede ser a causa de falta de diagnóstico oportuno y estudios genéticos. Adicionalmente, se encontró que las variantes aisladas en los pacientes sobre el gen LDLR (1187-1 G>T y 1187-1 G>A) se reconocen como causa de hipercolesterolemia familiar 6.
En el estudio de Chmara en Polonia se reportó por primera vez la variante 1187-1 G>T, variante que padece la familia comentada en la serie de casos 7.
En el estudio de López en Colombia, por su parte, se encontraron tres mutaciones identificadas como patogénicas: la variante a c.11G > A, la n c.416A > G y la c.1187G > A. 8.
A diferencia de los resultados encontrados en el estudio de López, la mutación de 1187-1 G>T en la serie de casos es clasificada como patogénica, mientras que la mutación de c.1187G > A es clasificada como de significado clínico incierto. Sin embargo, en el estudio realizado en Polonia se describen ambas variantes como patogénicas 7,8. Tanto en los estudios encontrados como en el caso presentado, ambas variantes se presentan simultáneamente en todos los pacientes, por lo que podría haber una estrecha relación entre una mutación y otra. Sin embargo, se requieren estudios de extensión para confirmar esta relación. De esta posible relación se genera también la interrogante acerca de cuál de ellas es la principal causante del fenotipo de la enfermedad.
Las manifestaciones clínicas de la enfermedad son importantes para considerar tanto la sospecha de hipercolesterolemia familiar como para la correlación de las mismas a desenlaces clínicos específicos. Se ha demostrado que la presencia del xantoma del tendón de Aquiles, como en este caso, es un signo de exposición a largo plazo al colesterol alto en sangre y mayores concentraciones de LDL-C 9. Otro estudio prospectivo demostró que aquellos pacientes con síndrome coronario agudo, especialmente con elevación del ST, tenían mayor prevalencia de xantoma del tendón de aquiles 10. El arco corneal, por su parte, es otro hallazgo al examen físico presente en la paciente que además se correlaciona con la evolución de la enfermedad y puede ser útil para predecir desenlaces clínicos determinados 11.
La disminución de niveles de C-LDL es importante, ya que este tipo de pacientes cuenta con un riesgo muy alto de desarrollar enfermedades cardiovasculares. La meta consiste en mantener niveles de LDL por debajo de los 100-130 mg/dl. Existen pacientes a quienes es difícil aplicar esta meta. Sin embargo, se ha comprobado que una reducción de LDL del 50 % genera una disminución notoria en el grosor de la capa íntima-media de la carótida 12.
Por otra parte, es importante resaltar el diagnóstico oportuno de enfermedades con baja prevalencia como la hipercolesterolemia familiar. El "tamizaje en cascada" es uno de los métodos diagnósticos más costo-efectivos ante estas situaciones, dado que no solo se diagnóstica al paciente en sospecha, sino también a los miembros de su grupo familiar pertinentes, lo cual ha demostrado una disminución del tiempo entre el diagnóstico e inicio del tratamiento 13.
Cabe resaltar la importancia de la asesoría genética para la hipercolesterolemia familiar. Es la única causa genética de enfermedad coronaria prematura, para la cual un enfoque sistemático basado en la población para encontrar a las personas afectadas y evaluar a sus familias está claramente justificado en este momento 14. Siendo la causa hereditaria más común de enfermedad coronaria prematura, conduce a una morbimortalidad significativa, por lo que los asesores genéticos, al ser profesionales de la salud especializados en capacitación tanto en genética médica como en asesoramiento psicosocial, cumplen un rol importante tanto para el diagnóstico como para el seguimiento de estos pacientes. Los pacientes con HF y sus familias requieren educación enfocada sobre la naturaleza hereditaria de su enfermedad, el riesgo para los miembros de la familia, la necesidad de la detección en cascada y la disponibilidad de pruebas genéticas 15,16.
CONCLUSIONES
La hipercolesterolemia familiar (HF) es una patología de origen genético que conlleva a alteraciones multisistémicas y aumenta el riesgo de enfermedad cardiovascular a corta edad, así como genera cambios en la calidad de vida del paciente y su familia.
Presentamos el caso de cuatro pacientes que padecen la mutación, de diferentes edades, quienes pertenecen a la misma familia y la mayoría de ellos son tratados con terapia farmacológica. Es importante resaltar la importancia de realizar un tamizaje familiar oportuno para disminuir los riesgos y evitar que aumente la afectación a múltiples órganos.
Por otro lado, los casos de HF no han sido registrados en Colombia y es pertinente el conocimiento de la mutación por el personal médico, para así saber abordarlo adecuadamente y generar un diagnóstico temprano antes que genere riesgos importantes y cambie la calidad de vida de los pacientes y su entorno.
El tratamiento de primera línea de la hipercolesterolemia se basa en estatinas en sus dosis máximas posterior al diagnóstico. Adicionalmente, vigilar la tolerancia y aparición de efectos adversos. Así como se recomienda una reducción de al menos el 50 % del LDL.
Finalmente, se debe mencionar las dificultades que padecen los pacientes en nuestro país con respecto al acceso al sistema de salud, en especial con las diferentes enfermedades genéticas, que en muchas ocasiones son desconocidas y no se diagnostican de forma oportuna.

