











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
Introduction
Within asymmetric catalysis, styrene and α-methylstyrene are found as substrates of great interest since their enantiomeric epoxides have highly desirable properties due to their reactive versatility for which they are involved in the synthesis of medicines and agrochemicals (Wong & Shi, 2008; Xia, et al., 2005). Although these substrates have been widely used (Huang, et al., 2019; Tang, et al., 2008) the chromatographic methods for the quantification of styrene and α-methylstyrene in this type of reaction have not been validated (Berijani, et al., 2019; Hadian & Hosseini, 2016) and, therefore, the results have not been supported. In this context, the objective of the present study was to report the results of the validation of a chromatographic method for the quantification of these olefins in samples and/or enantioselective epoxidation tests. The method we describe here for styrene has comparable results with those reported in other areas (Gennari, et al., 2012) as regards the limits of detection (LOD) and quantification (LOQ) and it is the only validated method of GC-FID quantification of α-methylstyrene. Additionally, the method also works with an FID detector, which was not the case with a previously reported MS detector (Bubeníková, et al., 2019; Even, et al., 2019; Hwang, et al., 2019), and it allows simultaneous quantification of the two substrates thus expanding the possible fields of application.
Materials and methods
Materials
We developed and verified the method using commercial samples of styrene and α-methylstyrene (Sigma-Aldrich, St. Louis, United States) and toluene (Merck, Darmstadt, Germany). We separated the analytes on a chiral capillary column (β-DEX, Sigma-Aldrich, St. Louis, United States) of 30 m in length, 0.25 mm of internal diameter, and a 0.25 film thickness, located on a Varian-CP 3800 gas chromatograph (Palo Alto, United States) with a flame ionization detector (FID). The method was developed under the following conditions: injector temperature, 280°C; split ratio, 1/100; injection volume, 0.2 µL; oven heating program, 50°C for 1 min at 2°C/min, 80°C for 2 min at 10°C/min, and 200°C for 12 min; FID detector temperature, 280°C. We used helium as the carrier gas at a flow and head pressure of 30 ml/min and 33 psi and the analysis time was 30 min.
We estimated the areas under the curve using the Interactive Graphics software of the Varian MS Workstation version 6.6®.
Verification of the chromatographic method
To verify the chromatographic method we validated the quality parameters of selectivity, linearity, LOD, LOQ, precision, and accuracy. Following the steps of a typical experiment, we added 2 ml of dichloromethane to the amount required according to the calibration curve under evaluation (3.3 - 19.6 µl styrene and 3.7 - 22.1 µl α-methylstyrene); then we diluted an aliquot of 100 µl in 2 ml with the same solvent, and, finally, we added 15 µl of toluene as the internal standard. We injected the resulting sample under the conditions previously mentioned (Table 1) and we collected the chromatographic area and elution time data.

Selectivity. Selectivity refers to the ability of the method to measure and/or identify simultaneously or separately the analytes of interest (Aguirre, et al., 2001; Nageswara, 2018). Using the selectivity factor α as in Ec. 1, we determined if the compounds evaluated were chemically distinct under the proposed method:

where t r A and t r B are the retention times of styrene and α-methylstyrene, respectively, and t m is the dead time of the analysis.
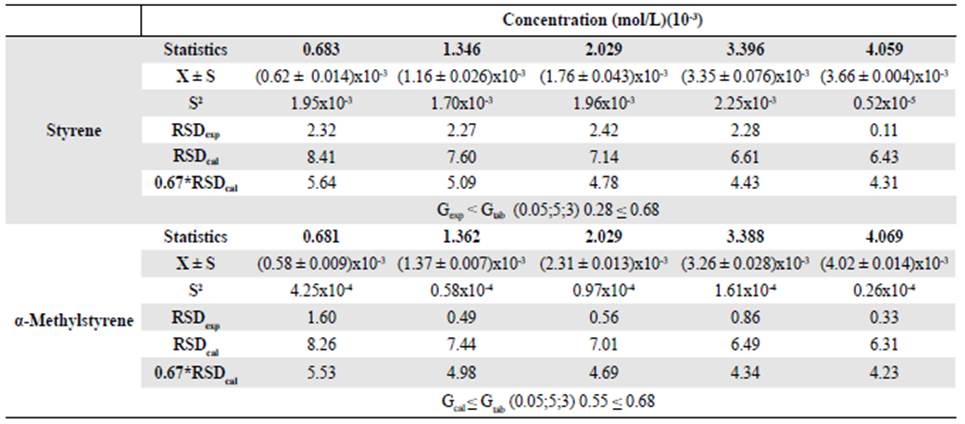
Linearity. The linearity defines the ability of a method to obtain test results propor tional to the analyte concentration in a sample (UNODC, 2009). In gas chromatography, the calibration with an internal standard is very often used (Poole, 2012). Here we deter mined the linearity of the method by constructing a calibration curve with five levels of concentration of the commercial sample injected in triplicate. As acceptance criteria, we used various parameters of linear regression: A correlation coefficient (r) ≥ 0.99, a determination coefficient (r2) ≥ 0.98, a slope significantly different from 0 (texp > tcal), and a Cochran test (Gexp < Gtab) as proof of the homogeneity of variances.
Limits of detection and quantification. The LOD of an analytical procedure is defined as the lowest detectable amount of analyte in a sample, that may not necessarily be quantifiable as an exact value (ICH, 2005; Gabhe, 2015). On the other hand, the LOQ is the lowest amount of analyte in a sample that can be quantitatively determined with the appropriate precision and accuracy (ICH, 2005; Gabhe, 2015). According to the ICH and IUPAC guidelines, the detection limit can be determined from the standard deviation of the replicated blank measurements and the slope of the calibration curve. However, when it is difficult to determine them due to instrumental noise, they can be calculated from enriched samples with a concentration close to zero.
We calculated LOD (X d ) and LOQ (X c ) using Ec. 2, 3, and 4 (Poole, 2012), where b 1 is the slope of the calibration curve, σ bl is the standard deviation of the concentration sample close to 0, and k c , k d y k q are statistics constants that depend on the level of significance (α) and the degrees of freedom (gl = n-1). In this case, α = 0.05 y gl = 9, so that k c = k d = 1.83, y k q = 10.



Precision. The precision of an analytical method represents the proximity between the series of measurements obtained from the same sample under the prescribed conditions (FDA, 2001). Repeatability is the estimated precision in the most optimal operating conditions: The same laboratory, the same analyst, the same instrument, and a short time interval. To assess the precision of the method, we injected each level of the calibration curve three times. We determined the experimental relative standard deviation (RSDexp) and we compared the value obtained to that of the standard deviation (RSDcal) in Horwitzs equation (Ec. 5).

Where C is the concentration of compound expressed as a dimensionless mass fraction at each level of the calibration curve. The precision is considered acceptable when the RSDexp is less than 0.67*RSDcal (Zuas, et al., 2016). We used a Cochran test, where the calculated statistic should be less than the tabulated one, to determine if the concentration had an effect on precision.
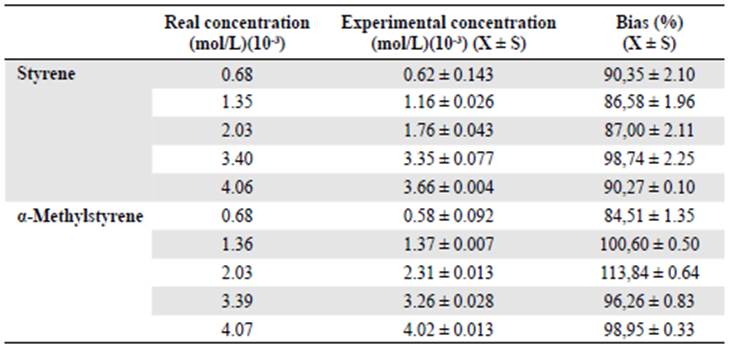
Accuracy. Accuracy is the proximity between the average value of a number of test results and the accepted reference value (Poole, 2012). We evaluated this parameter through the percentage of recovery or bias (Ec. 6).

where x is the average of the experimental concentrations of three injections for each level of the calibration curve found through the equation and x ref is the real concentration used. Commonly accepted values are between 100±20% (FDA, 2001, Gomes, et al., 2010).
Results and discussion
Selectivity
Given the retention times of 5.47 min for styrene (A) and 9.38 min for α-methylstyrene (B) and a dead time of 1.02 min, the selectivity factor of B with respect to A equaled 1.87 indicating that under the working conditions the chromatographic method exhibited selectivity towards the two compounds.
Linearity
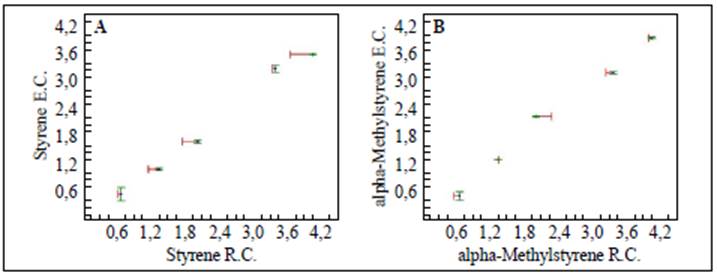
The results presented in figure 1A and B and in table 1 indicated that the method was linear for both substrates in the concentration range evaluated since we obtained correlation coefficients (r) over 0.99 and determination coefficients (r2) over 0.98. On the other hand, texp > ttab and Gexp < Gtab evidenced that the slope was significantly different from 0 and that the concentration had no effect on the linearity of the method, respectively.

Limits of detection and quantification
In table 2 we summarize the values of LOD and LOQ found for both styrene and α-methylstyrene. These results showed that the chromatographic method had remarkably low values for the quantification of these substrates.

Precision
The results shown in table 3 indicated that the analytical method met the accepted criteria of repeatability since at all the concentration levels evaluated the RSDexp was less than 0.67*RSDcal. In its turn, the concentration variable had no effect on the precision since Gcal ≤ Gtab.
As shown in table 4 and figure 2A-B, the bias values or recovery percentage at all concen tration levels for the two olefins used were within the acceptable values for this parameter (100±20%), which means the method was accurate throughout the range of work.
Conclusions
The chromatographic method developed for the quantification of styrene and α-methylstyrene had acceptable results for each validation parameter and providing sufficient evidence to assert that it is a reliable, selective, linear, precise, and accurate method in the concentration ranges evaluated. The method and the enantioselective epoxidation reactions can be used for the analysis of samples containing either of the two substrates in plasticizers, resins, or polymers that commonly involve them.

