











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
Introducción
El síndrome hemolítico urémico (SHU) atípico (SHUa) es una enfermedad grave y huérfana que se presenta en el 10 % de los pacientes con SHU y puede ser de origen familiar o esporádico (1, 2). Aproximadamente en un 50 % de los casos se detectan mutaciones que afectan a las proteínas reguladoras de la vía del complemento, llevando a una sobreactivación no controlada de la misma (2-4). La proteína cofactor de membrana (MCP) es un regulador transmembrana que actúa como cofactor para el cofactor I (CFI) (1) e inactiva C3b en la superficie de las células endoteliales (3). Se han descrito alrededor de 30 mutaciones diferentes en el gen que codifica para MCP (1, 5, 6), las cuales solo se presentan en el 10-15 % de pacientes con SHUa (1, 7, 8).
Las manifestaciones clínicas del SHUa son variables, documentándose con mayor frecuencia las de tipo renal. Si bien la desregulación de la vía alterna del complemento puede manifestarse en otros órganos, en menos del 50 % de los pacientes se han descrito manifestaciones extrarrenales, dentro de las cuales podemos encontrar compromiso de sistema nervioso central, cardiovascular, pulmonar, cutáneo, retiniano y gastrointestinal (9). La literatura actual describe que entre el 8,5 y el 12 % de los pacientes con SHUa cursan con afectación del sistema gastrointestinal, pero solo se ha descrito pancreatitis en el 1,7 % y elevación de transaminasas en el 1,1 % de los enfermos (9). Estos pacientes usualmente presentan una mayor tasa de recaídas y mayor mortalidad, lo que implica un pobre pronóstico (1).
Reportamos un caso de un paciente de 8 años que cursó con SHUa con compromiso extrarrenal hepatopancreático y hematológico, asociado a afectación renal, en quien se identificó una mutación en el codón de inicio de MCP probablemente patogénica, no reportada en la literatura. El paciente fue manejado con eculizumab con adecuada tolerancia.
Presentación del caso
Paciente masculino de 8 años con cuadro clínico de tres días de fiebre, asociado a dolor abdominal tipo cólico intermitente y dolor en región lumbar, deposiciones líquidas sin moco ni sangre, emesis, hematemesis, astenia, adinamia, malestar general y hematuria. Sin antecedentes patológicos, recién nacido a término, adecuado peso y talla al nacer, sin hospitalizaciones previas, sin noción de contagio, inmunización al día, sin cirugías ni traumas previos, sin antecedentes de transfusiones ni antecedentes familiares. Fue manejado en casa con acetaminofén vía oral sin mejoría.
El paciente ingresó deshidratado, ictérico y con dolor abdominal, por lo que inicialmente recibió manejo con líquidos endovenosos. Recibió bolo de solución salina normal de 20 cc/kg y se continuaron a 2000 cc/m2/día, con posterior aumento a 2500 cc/m2/día.
Durante la hospitalización presentó signos de sobrecarga hídrica e hipertensión severa estadio II. Se documentó lesión renal aguda KDIGO 3 con anuria y creatinina pico en 2,6 mg/dL, sin respuesta a cristaloides y diurético de asa (furosemida en infusión continua a 0,1 mg/kg/hora durante cuatro días, posteriormente a 1,6 mg/kg/día durante tres días y finalmente a 1,2 mg/kg/día), por lo que requirió inicio de terapia de reemplazo renal y fue trasladado a cuidado intensivo pediátrico (UCIP). Presentó además anemia y trombocitopenia severa (figura 1), extendido de sangre periférica con linfopenia absoluta y esquistocitos, hiperbilirrubinemia, deshidrogenasa láctica (LDH) elevada, Coombs directo negativo, tiempos de coagulación sin alteraciones, complemento C3 bajo y C4 normal, requiriendo múltiple soporte transfusional con plaquetas y glóbulos rojos. Se documentó elevación de aminotransferasas, amilasa y lipasa, evidenciando compromiso hepatopancreático, por lo que recibió manejo para pancreatitis aguda.
Ante el cuadro clínico descrito se diagnosticó microangiopatía trombótica (MAT) aguda con compromiso multiorgánico (gastrointestinal severo, hematológico y renal), por lo que se sospechó de probable SHUa como causa, ante lo que se iniciaron estudios complementarios y terapia con recambios plasmáticos, sin obtener mejoría en el cuadro clínico y con persistencia de trombocitopenia severa. Se descartó púrpura trombocitopénica trombótica (PTT) con reporte de ADAMTS13 normal (86 %), coagulación intravascular diseminada (CID) y síndrome antifosfolípidos (SAF), con serología negativa, hemocultivos negativos y reporte de toxina Shiga negativa.
Previa vacunación para meningococo y neumococo, se inició manejo con eculizumab en dosis de inducción de 600 mg y posteriormente dosis de mantenimiento de 600 mg a los siete días de la primera. Documentando una adecuada respuesta clínica, dado que presentó mejoría del recuento plaquetario a las 24 horas de la primera dosis, además de estabilización de la hemoglobina, recuperación de función renal y hepatopancreática, mejoría del volumen urinario, normalización de presión arterial y resolución del dolor abdominal, logrando suspender la terapia dialítica sin nuevas manifestaciones de sangrado ni hemólisis. La variación en los exámenes paraclínicos se ilustra en la figura 1 y en la tabla 1.
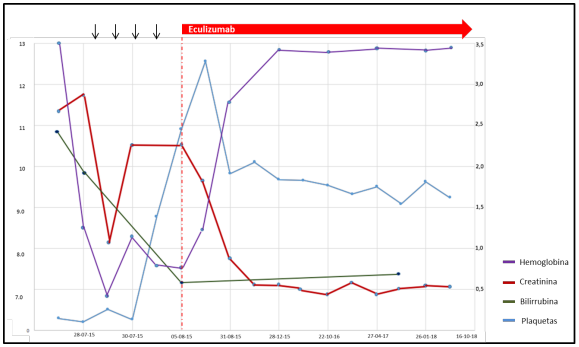
Fuente: elaboración propia.
Figura 1 Variación en exámenes paraclínicos del paciente. Nota aclaratoria: se observa la respuesta del paciente a las diferentes terapias instauradas, con flecha se identifican las sesiones de recambio plasmático terapéutico y en rojo el inicio y respuesta al tratamiento con Eculizumab.
Tablas complementarias a la figura 1
Valores de hemoglobina (g/dL)

Valores de bilirrubinas (µmol/L)

Valores de plaquetas (celulas/mL)

Valores de linfocitos (células/mL)

Valores del complemento (mg/dL)

Valores de transaminasas (UI/L)

Valores de amilasa (UI/L)

Valores de lipasa (UI/L)

Valores de LDH (UI/L)

Tabla 1 Paraclínicos del caso

Nota aclaratoria: entre paréntesis están los valores de referencia.
Fuente: elaboración propia.
Al paciente se le realizó un estudio molecular de los genes asociados a SHUa, el cual reportó la presencia de una mutación heterocigota c.1a>G (p.Met?) en el gen MCP, clasificada como probablemente patogénica. El paciente recibió manejo con eculizumab por tres años con remisión completa, asintomático, sin nuevas recaídas, por lo cual, en junta multidisciplinaria (Nefrología, Nefropediatría y Genética) y considerando a partir de la literatura actualmente disponible con la mutación documentada, hay un buen pronóstico a largo plazo a pesar de una alta probabilidad de recaída, por lo cual se suspendió tratamiento con eculizumab y actualmente el paciente continúa con buena función renal sin recidivas (función renal postratamiento: creatinina 0,51 mg/dL, TFG 135 ml/min/1,73m2, creatinuria 96,7 mg/dL, índice microalbuminuria/creatinuria 2,5 e índice proteinuria/creatinuria 0,12).
Discusión
El SHUa es un trastorno complejo multigénico que se caracteriza por la alteración de la vía alterna del complemento, genera daño endotelial y clínicamente se manifiesta como microangiopatía trombótica, además usualmente tiene un curso recidivante con alto riesgo de enfermedad renal crónica avanzada (9-12). Han sido identificadas deficiencias hereditarias o adquiridas en la vía alterna del complemento y hay datos de Francia para el periodo 2000-2008 donde el SHUa tiene una incidencia de 0,23 por millón por año, dentro de los cuales el 61 % presentaba mutaciones en genes del complemento (8, 13).
La literatura incluye muchos informes sobre el compromiso renal de los pacientes con SHUa y su desenlace, pero solo datos limitados sobre la afectación en la microvasculatura extrarrenal (9, 14). Se conoce que estas últimas ocurren en el 20 % de los casos, especialmente en la fase de presentación inicial de la enfermedad, pero podrían ocurrir incluso después de años del debut (9, 15, 16) y se ha descrito que estas pueden ser potencialmente mortales (17). En un estudio del 2018 que evaluó los registros turcos y europeos de SHUa, se encontró que la manifestación extrarrenal más común había sido la afectación del sistema nervioso central (27,2 %), seguida del compromiso gastrointestinal (11,8 %), cardiovascular (7 %) y respiratorio (7 %); sin embargo, ninguno de estos pacientes tenía mutaciones en el gen MCP (9).
Un reciente estudio canadiense documentó que las manifestaciones gastrointestinales son más comunes en adultos que en niños (70 % versus 50 % respectivamente), siendo más frecuentes en el SHUa en presencia de anticuerpos del factor H (15, 17). Dentro de estas se ha descrito que la afectación más común es la elevación de enzimas pancreáticas y el daño hepático (16, 17), sin embargo, existen datos de que la pancreatitis se presenta solamente en el 1,7-8,5 % de los pacientes y la elevación de transaminasas en el 1,1-8,5 % de los casos (9).
Se ha descrito que el daño microangiopático intrapancreático es secundario a isquemia tisular pancreática, que puede contribuir al síntoma más común de dolor abdominal, pero se desconoce la fisiopatología exacta del desarrollo de la pancreatitis y se ha considerado que el daño esté relacionado con la microangiopatía trombótica como la causa de la disfunción de las células endoteliales. Además, se planteó que la pancreatitis puede ser la primera manifestación de un episodio agudo de SHU, pero también se ha descrito como un desencadenante de la aparición o recurrencia de la MAT (16). Lo anterior se podría explicar en que la respuesta inflamatoria aguda a la pancreatitis (mediada por factor de necrosis tumoral alfa, interleucina (IL) 1, IL-6, IL-8, otras citocinas y proteasas pancreáticas) pueden conducir a la activación endotelial, daño de células endoteliales, activación de la coagulación intravascular y liberación del factor de Von Willebrand (vWF), agravando los procesos de MAT sobre la base de predisposiciones potenciales (incluidos los defectos de las proteínas reguladores del complemento) (16).
Asimismo, la afectación del páncreas se puede presentar en el 3-15 % de los pacientes con SHU-shigatoxina y puede llegar a generar disfunción significativa de las células beta, llevando al desarrollo de diabetes mellitus insulinodependiente, incluso años después de la presentación del SHU (16).
El riesgo de recurrencia depende de la variante genética, siendo del 50-80 % en las que presentan mutaciones patogénicas en genes que codifican para CFH, CFI, factor B o C3; en contraste con el 8% de recurrencia en la variante de MCP y del 8-28 % en pacientes sin una mutación identificada (18); sin embargo, el riesgo de recaída es mayor en pacientes con SHUa con mutaciones en MCP que debuta en la infancia (14, 19), esto en comparación con el estudio francés donde describen una frecuencia de recaída después de un año del 92 % en niños con mutación del gen MCP y aproximadamente del 30 % en el resto de los subgrupos (13). Asimismo, documentaron mayor mortalidad en niños que en adultos (6,7 % vs. 0,8 % a un año) (13).
Presentamos el caso de un paciente con SHUa con una mutación de novo en el gen MCP que afecta el codón de inicio de la traducción, con manifestación predominante de tipo hepatopancreático severo, asociación que ha sido poco reportada previamente en la literatura. Otro cambio nucleotídico en la misma posición ha sido descrito como causal de SHUa en otros pacientes (1). Entre el 10 y el 20 % de los pacientes con SHUa cursan con mutaciones en MCP, la mayoría son de tipo missense, pero también se han reportado nonsense y variaciones en el sitio de splicing (20). Más del 75 % de las mutaciones reportadas causan una reducción en la expresión de MCP y se encuentran generalmente de manera heterocigota (21).
La edad promedio de SHUa es de 8,5 años (rango 5-13 años), como en nuestro paciente, con tasas de recaídas del 60 % y una tasa de mortalidad o progresión a enfermedad renal crónica avanzada del 20 % (1, 13). Los pacientes con mutaciones en el gen MCP cursan con un inicio más temprano de la enfermedad que pacientes con CFH, CFI o que no tienen mutación identificada. La enfermedad es de penetrancia incompleta, con tasas de penetrancia del 54 % (8). Los valores séricos de complemento C3 y C4 pueden encontrarse por debajo del nivel normal, no obstante, este hallazgo se presenta solo en 30-50 % de los casos, principalmente con niveles bajos de C3 en pacientes con mutaciones en CFH, MCP, CFI y trombomodulina o con anticuerpos anti CFH (1, 22).
En el estudio de Sheerin et al., uno de los pacientes con SHUa con una mutación conocida en CD46 (c.646T>G; p.Trp216Gly) se presentó con episodios recurrentes de pancreatitis, similar al caso que presentamos (4). En el caso de este paciente, el inicio de eculizumab no redujo la frecuencia de pancreatitis y este medicamento fue suspendido luego de 15 semanas de tratamiento, a diferencia de nuestro caso que resolvió la pancreatitis posterior al inicio de eculizumab.
Pacientes pediátricos con mutaciones MCP tienen buen pronóstico renal a largo plazo, con un 25 % del riesgo de progresión a enfermedad renal crónica avanzada (seguimiento promedio de 17,8 años). La frecuencia de recaídas después de un año fue de 92 % en niños con SHU asociado a MCP y de 19-50 % en otros subgrupos. El buen pronóstico renal relacionado con mutaciones en MCP parece ser cierto solamente si el primer episodio de la enfermedad ocurrió durante la infancia.
En esta cohorte, el pronóstico de SHUa sin mutación identificada y asociado a MCP fue similar en niños (13). En el caso de nuestro paciente, este lleva dos años de seguimiento posterior a suspender el tratamiento con eculizumab sin presentar recaídas y con una función renal completamente normal.
Un cambio en la historia natural de la enfermedad se generó con la introducción de eculizumab en el manejo de SHUa, un anticuerpo monoclonal que se une específicamente a la porción terminal del complemento C5 y bloquea el clivaje de C5-C5a y C5b, inhibiendo la progresión de la vía terminal de complemento al complejo de ataque de membrana (6, 22). En estudios prospectivos, el tratamiento con eculizumab ha disminuido el riesgo de progresión a enfermedad renal crónica avanzada del 60-70 % al 15-20 % en adultos y del 30-40 % al 5-10 % en niños (23).
El estudio molecular de los pacientes es fundamental en SHUa, ya que permite predecir el curso natural de la enfermedad, el riesgo de presentar complicaciones extrarrenales y potencialmente determinar la duración de tratamiento y prevención de recurrencia postrasplante. De igual forma es importante que estos pacientes y sus familias reciban asesoramiento genético apropiado, seguimiento multidisciplinario, detección precoz de recaídas y manejo con eculizumab para disminuir riesgo de daño renal y sistémico permanente y severo.
Conclusiones
Es mucha la evidencia y literatura que existe actualmente alrededor de esta patología y el compromiso sistémico documentado está claramente explicado desde el punto de vista fisiopatológico, sin embargo, el debut con manifestaciones extrarrenales suele ser una presentación atípica de la enfermedad y aun así ser la clínica predominante que le permita al médico sospechar el diagnóstico, por lo que se vuelve imperativo dar a conocer y educar a la comunidad científica alrededor de los signos y síntomas menos frecuentes que pueden presentar estos pacientes, con el fin de evitar retrasos en el diagnóstico y la oportunidad de manejo adecuado, evitando daños irreversibles y, en últimas, lograr mejorar la sobrevida y pronóstico de la población pediátrica.
Por lo anterior, se espera que con este caso clínico se fomente el interés por aumentar el conocimiento y la educación de los profesionales de salud sobre esta patología. Además de incentivar la realización de futuros estudios que permitan el correcto seguimiento a largo plazo en estos pacientes y que determinen el impacto de retirar el tratamiento en la calidad de vida y recaídas.

