











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkLos avances científicos producidos en la segunda mitad del siglo xx han sido claves para el desarrollo de los medicamentos biotecnológicos. En el año 1950 se produce el punto de inflexión para llegar al descubrimiento de los biotecnológicos, cuando los científicos James Watson y Francis Crick, presentan el hallazgo de la estructura molecular del ADN, molécula portadora de la información e instrucciones genéticas de los organismos vivos.
En 1972, Berg, Cohen y Boyer introducen la tecnología del ADN recombinante. Y en la primera mitad de los años setenta, Georges Köhler junto con César Milstein y Niels Jerne descubren los anticuerpos monoclonales, en el laboratorio de biología molecular de Cambridge (Reino Unido). mAbs es la nomenclatura adoptada tanto por la United States Adopted Names (USAN) de Estados Unidos, como por la Denominación Común Internacional (DCI) de la Organización Mundial de la Salud (OMS) para los productos farmacéuticos.
Estos autores investigaban los mecanismos moleculares de la generación de diversidad de los anticuerpos y necesitaban producir una célula B inmortal con especificidad conocida, para así poder analizar en detalle las mutaciones de los genes de las inmunoglobulinas (Ig). Para ello fusionaron una línea de células de mieloma murino, capaz de multiplicarse indefinidamente, con células de bazo de un animal inmunizado, de corta vida, pero productoras de anticuerpos determinados. Cultivando estas hibridomas en medio selectivo, lograron seleccionar células con ambas características: una línea inmortal productora de anticuerpos de especificidad previamente determinada.
El 7 de agosto de 1975 Köhler y Milstein publicaron en la revista Nature el artículo "Continuous cultures of fused cells secreting antibody of predefined specificity", el cual les valdría el Premio Nobel de Medicina y Fisiología nueve años más tarde. Los científicos habían inventado la técnica para fabricar anticuerpos monoclonales con un enorme impacto en la medicina y la ciencia biológica básica, hecho que catalizó muchos otros descubrimientos 1.
Finalmente, el primer uso terapéutico en humanos tuvo lugar en 1982 para el tratamiento de un linfoma. Y en 1986 se comercializa el primer mAbs, MuromonabcD3 para minimizar el rechazo en pacientes trasplantados.
Independientemente del uso de los anticuerpos monoclonales en técnicas de diagnóstico, que han supuesto una revolución en el campo de la histopatología y permitido el desarrollo de diversas técnicas de laboratorio como la citometría de flujo o la inmunofluorescencia, entre otras, las posibilidades de aplicación para tratar enfermedades humanas son amplísimas, siendo la oncología el área de aplicación terapéutica más importante 2.
MÉTODO
Se realizó una búsqueda bibliográfica en revistas indexadas, con un enfoque analítico y retrospectivo. Se realizó una búsqueda de artículos en las principales bases de datos bibliográficas disponibles en Internet, entre ellas Dialnet, SCÍELO, Science Research, Latindex, Scopus, GaBijorunal, PubMed, ResearchGate, Elsevier, AHA Journals RSS, Open Journal Systems, International Journal of Science and Research (IJSR), Google Academics. Para la búsqueda más compleja, que fue aquella realizada en PubMed/MEDLiNE, se utilizaron descriptores MesH.
Se excluyeron trabajos de revisión, editoriales y comunicaciones a congresos. En caso de encontrar un artículo repetido en varias publicaciones se incluyó aquel publicado en una revista con mayor factor de impacto y/o aquel publicado más recientemente. Se encontraron 57 artículos y se utilizaron las 23 publicaciones que figuran en las referencias bibliográficas.
El objetivo general es una revisión bibliográfica de artículos científicos con la intención de hacer una descripción de la evolución en el uso de los anticuerpos monoclonales en el tratamiento del cáncer, y aportar una visión económico-sociosanitaria de la obtención, producción y comercialización de las nuevas terapias.
El objetivo específico es determinar el justo medio entre los tiempos de vigencia de patentes exclusivas y el acceso a los tratamientos por parte de los sistemas de salud, y los enfermos de cáncer.
Anticuerpos terapéuticos
Los anticuerpos o inmunoglobulinas (Ig) son glicoproteínas producidas por el sistema inmune de los vertebrados que, gracias a su capacidad natural de enlazarse de manera específica a una gran diversidad de moléculas 3, cumplen la función de identificar y neutralizar agentes ajenos al organismo como bacterias o virus (antígenos). Son producidos por los linfocitos B (células sanguíneas) los cuales, una vez expuestos a estos blancos o antígenos, se activan y diferencian a células plasmáticas productoras de anticuerpos específicos contra este antígeno.
Los anticuerpos pueden encontrarse tanto de manera soluble (libres en sangre) como anclados en la membrana de linfocitos B (linfocitos B de memoria).
Estructuralmente están formados por dos cadenas polipeptídicas pesadas H (del término anglosajón heavy, 'pesado') y dos cadenas ligeras L (del término anglosajón light, 'ligero') unidas entre sí mediante enlaces covalentes. Las cadenas ligeras consisten en una región variable (VL) y una constante (CL) mientras que las pesadas presentan una región variable (VH) y tres constantes (CH1, CH2, CH3). Funcionalmente podemos distinguir dos porciones en los anticuerpos: una de ellas implicada en el reconocimiento y unión al antígeno, denominada región Fab (antigen binding Fragment) y otra implicada en la modulación de mecanismos de defensa mediante la interacción con proteínas y células inmunes, llamada región Fc (crystallizable Fragment) (Figura 1).
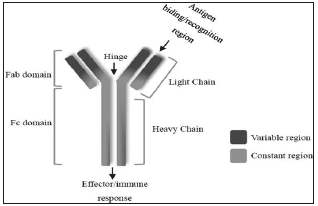
Fuente: Calvo G.Lens C, Sanz J, de Mora F, Dorrego A, Hernández C, et al. El libro Blanco en los medicamentos biosimiilares en España; Innovación y Sostenibilidad, Fundación Gaspar nasal, 2017.
Figura 1 Estructura de un anticuerpo monoclonal
Existen cinco clases diferentes de anticuerpos que se diferencian entre sí por una serie de cambios estructurales que les confieren diferentes funciones en el organismo. Los cinco tipos de anticuerpos se denominan: Ig G, Ig M, Ig E, Ig A e Ig D 4.
Mientras que la técnica de la hibridoma ha permitido producir anticuerpos monoclonales, la tecnología del ADN recombinante otorgó la posibilidad de modificar estos anticuerpos con el fin de mejorar su eficacia, como puede ser la combinación de secuencias genéticas procedentes de diferentes organismos.
Los anticuerpos monoclonales, según su origen y estructura, pueden clasificarse en murinos (provenientes de ratón), quiméricos (en los que una porción de la proteína es humana y la otra de origen murino), humanizados (en los que casi toda la molécula es humana y solo pequeños fragmentos de la secuencia de aminoácidos proviene de otra especie) o enteramente humanos. Este proceso progresivo de humanización de los anticuerpos monoclonales responde a la necesidad de reducir su inmunogenicidad, y así poder usarlos como herramientas en procedimientos diagnósticos, en profilaxis y en terapia en humanos 5.
La quimerización se desarrolló a partir de 1984. Mediante la técnica de humanización, en 1986 se obtuvieron mAbs humanizados donde solo las regiones hipervariables de las cadenas ligeras y pesadas son de origen murino.
Un nuevo enfoque radicalmente diferente para abordar el problema de la humanización de los anticuerpos es la generación de hibridomas de ratón, que producen anticuerpos totalmente humanos. Es el caso de las lg procedentes de ratones transgénicos, a los cuales se les han reemplazado los genes de las regiones variables propias por las humanas, en las que los ratones llevan a cabo la recombinación de los genes VDJ, responsables de la codificación y ensamblaje de las lg. Los anticuerpos obtenidos tienen una alta afinidad con secuencias terminales humanas 6.
El empleo de anticuerpos humanizados y humanos ha mejorado notablemente su tolerancia clínica. Y la manipulación de los anticuerpos mediante la unión a otras moléculas o el diseño de nuevos fragmentos abren un gran abanico de posibles aplicaciones en medicina.
Aplicación terapéutica
La oncología es, sin duda, el área de aplicación terapéutica más relevante. Los productos basados en anticuerpos monoclonales (mAbs) son altamente específicos para un antígeno particular. Las enfermedades autoinmunes son el grupo siguiente de patología humana en el que más se han empleado estos productos y, fundamentalmente, en artritis reumatoide, enfermedad inflamatoria intestinal, esclerosis múltiple, lupus eritematoso, así como en el rechazo de trasplantes, y enfermedad de injerto contra el huésped 7.
La terapia antineoplásica mediante los agentes quimioterapéuticos tradicionales se caracteriza por la toxicidad significativa a nivel de las células no tumorales, debido al estrecho margen terapéutico de la mayoría de estos fármacos. En cambio, en la terapia dirigida se utilizan inhibidores farmacológicos específicos, con el objetivo de aumentar los efectos antiproliferativos y citotóxicos sobre las células tumorales, así como disminuir los efectos colaterales que afectan los tejidos sanos. Una de las aproximaciones terapéuticas de mayor futuro en este sentido la constituyen los anticuerpos monoclonales.
Distintos laboratorios han producido mAbs con una respuesta específica a antígenos, expresados principalmente en la superficie de células cancerosas pero que están ausentes (o son expresados en menor concentración) en las células normales. La especificidad antigénica de los mAbs les permite estimular la respuesta inmune del huésped frente a las células tumorales, interferir en el crecimiento y diferenciación de las células tumorales mediante el bloqueo de factores de crecimiento y sus receptores, y la formación de inmunoconjugados con mayor actividad antitumoral mediante la unión a agentes citotóxicos, radioisótopos o toxinas 8.
En la actualidad, hay 14 medicamentos basados en mAbs que han sido aprobados para el tratamiento de pacientes con cáncer (Tabla 1).
Tabla 1 mAbs terapéuticos aprobados en Estados Unidos y por la Unión Europea para su uso en el tratamiento del cáncer
| Nombre | Anticuerpo | Antígeno objetivo | Área terapéutica | Aprobación por la FDA |
| Rituximab | Quimerico IgG1 | CD 20 | Linfoma de células B, NHL | 1997 |
| Leucemia linfocítica crónica | 2010 | |||
| Trastuzumab | Humanizado lg61 | HER-2 | Cáncer de mama metastásico | 1998 |
| Cáncer de mama temprano | 2010 | |||
| Cáncer de estómago metastásico | 2010 | |||
| Gentuzumab | Humanizado lgG1 | CD33 | Leucemia mieloide aguda | 2000 |
| Alezutumab | Humanizado lgG1 | CD52 | Leucemia mieloide crónica | 2001 |
| Ibritumomab | Mouse lgG1 conjugado a 90Y) | CD20 | NHL | 2002 |
| Tositumomab | Mouse lgG1conjugado a 131l | CD20 | NHL | 2003 |
| Bevacizumab | Humanizado lgG1 | VEGF | Cáncer de colon metastásico | 2004 |
| Non-small-cell lung cancer | 2006 | |||
| Cancer renal metastásico GBM | 2009 | |||
| Cáncer de ovario (en Europa solo) | 2011 | |||
| Cetuximab | Quimerico lgG1 | EGFR | Cáncer metastásico de colon | 2004 |
| Head and neck cancer | 2006 | |||
| Cáncer metastásico de colon(primer tratamiento) | 2012 | |||
| Panitumumab | Humanizado IgG2 | EGFR | Cáncer metastásico de colon | 2006 |
| Ofatumumab | Humanizado lgG1 | CD20 | Chronic lymphocytic leukaemia | 2009 |
| Removab | Bi-sepecific mouse/rat hibrid lgG | EpCAMX CD3 | Malignant ascites (in Area Therapeutic Europe) | 2009 |
| Ipilimumab | Humanizado lgG1 | CTLA-4 | Metastatic melanoma | 2011 |
| Brentuximab | Quimerico lgG1 | CD30 | ALCL and Hodgkin lynphoma | 2011 |
| Pertuzumab | Humanizado lgG1 | HER2 | Metastatic breast cancer | 2012 |
Fuente: Tomada de Anticuerpos monoclonales: desarrollo físico y perspectivas terapéuticas, Machado NP, Téllez, GA, Castaño JC.
Mercado de los mAbs
El impacto científico y tecnológico que han tenido los nuevos descubrimientos muestra especial relevancia en el mejoramiento de aplicaciones tecnológicas en el tratamiento del cáncer, una de las enfermedades más graves de nuestro tiempo debido a su elevada prevalencia, morbilidad y mortalidad 9.
Dentro del gran avance que supone el descubrimiento, desarrollo y aplicación de los mAbs, es de nuestro interés abordar el impacto económico y accesibilidad a las nuevas terapias.
El desarrollo de los anticuerpos monoclonales ha dado lugar a un gran número de patentes que, actualmente, están autorizadas y son comercializadas.
En el caso de medicamentos bajo patente, sin sustitutos o competidores próximos, y que tienen por lo tanto una posición de monopolio, la teoría económica predice que el precio se situará en el nivel en que se maximicen los beneficios, normalmente un precio muy superior al costo de producción directo. Por otra parte, la teoría predice que, si puede segmentar y aislar los mercados de su producto, el monopolista adoptará una estrategia discriminante, que consiste en fijar en cada mercado el precio que maximice los beneficios en dicho mercado. Esto puede dar lugar a que en un mismo país haya precios distintos en cada segmento 10. Esta es la situación de los anticuerpos monoclonales que se han transformado en un importante grupo de fármacos. Más de 40 anticuerpos terapéuticos están aprobados para terapia humana y cientos se encuentran en distintas fases de desarrollo y ensayos clínicos. Con la tasa de aprobación actual de unos cuatro productos por año, se estima que 70 productos de anticuerpos monoclonales estarán en el mercado para el año 2020, y las ventas mundiales combinadas serán de casi 125 mil millones de dólares 11.
Entre los años 2001 y 2002, el valor global del mercado de anticuerpos monoclonales aumentó un 37 % hasta la cifra de 5,4 millones de dólares. Los líderes del mercado fueron los anticuerpos quiméricos, con un crecimiento del 43 % y 3,8 miles de millones de dólares en ventas, seguidos de los humanizados, con más de 14 mil millones de dólares y un crecimiento del 29 % 12.
Hasta el presente, el modelo de negocio del sector de los anticuerpos terapéuticos ha evolucionado a través de tres rutas, financiación mediante capital de riesgo, colaboraciones de proyectos de I + D o licencias 13. Los costos de investigación y desarrollo (I + D), los costos de publicidad y comercialización y los relacionados con la gestión de la propiedad intelectual forman parte del financiamiento del sistema. Este tipo de costos no son imputables objetiva y unívocamente a un medicamento concreto, ni mucho menos a una unidad producida de un medicamento.
Así, por ejemplo, el gasto en todas las actividades de I + D, incluyendo tanto el de los proyectos que terminan con un producto más o menos exitoso en el mercado, como el de los proyectos fallidos en cualquier fase del proceso de I + D, deben ser recuperados a través de las ventas de los productos que tienen éxito comercial, cuyo precio debe permitir recuperar el gasto tanto de los proyectos exitosos como el de los que no lo fueron.
Esta realidad nos lleva a reflexionar sobre el equilibrio que debería lograrse entre la accesibilidad a los medicamentos de manera universal, y las patentes como títulos de propiedad intelectual destinados a recompensar y estimular la investigación y desarrollo de nuevos tratamientos.
El precio es una de las principales barreras de acceso a los medicamentos innovadores. Es un desafío su incorporación al Sistema Nacional de Salud con sostenibilidad, equidad y eficiencia. Otras barreras de acceso a los medicamentos biológicos y biosimilares que cumplen con las normas de la EMA, OMS y FDA pueden ser los pocos conocimientos y falta de comprensión de lo que son los medicamentos biológicos y su valor terapéutico por parte de los gobiernos, médicos y pacientes, la poca o ninguna regulación de los medicamentos biológicos y biosimilares, la escasa o ninguna voluntad política para garantizar el acceso 14.
El impacto de los biosimilares
Como se ha descripto en el capítulo anterior, la formación del precio de los medicamentos recientemente introducidos en el mercado está habitualmente protegida por derechos de exclusividad, y es muy distinta a la del resto de los medicamentos cuyos derechos de exclusividad han caducado y están, por tanto, sujetos a la competencia efectiva o potencial de proveedores genéricos.
Por ese motivo, es más que relevante el impacto de la caída de las patentes de anticuerpos terapéuticos que ha comenzado a darse en los últimos años, como es el caso de Rituximab (2013), Trastuzumab (2014), Etanercept (2015), Infliximab (2015), o que están próximos a su vencimiento, como es el caso de Adalimumab (2018), Bevacizumab (2022). Esta nueva realidad ha abierto la puerta al desarrollo de los denominados anticuerpos biosimilares.
Cuando hacemos referencia a un fármaco biosimilar, se trata de aquel medicamento biológico que se desarrolla para ser altamente similar a un medicamento biológico ya existente ("de referencia" u "original"), y que podrá comercializarse una vez que haya vencido la patente del medicamento de referencia 15.
Ya los anticuerpos monoclonales habían supuesto un cambio en el paradigma de tratamiento de muchas enfermedades, entre ellas oncológicas, inflamatorias y autoinmunes; a este cambio revolucionario se ha sumado un nuevo reto, la introducción de los anticuerpos monoclonales biosimilares que son producidos por un fabricante diferente al innovador, con una misma secuencia, sin ser idéntico al producto de referencia.
El coste de los biosimilares está asociado al tiempo que requiere su desarrollo. Si bien su costo es sensiblemente menor que el de un medicamento biológico original, no es comparable con la elaboración de un medicamento genérico. Mientras que el periodo de desarrollo promedio de un genérico ronda los tres años, tiempo necesario para demostrar su bioequivalencia, los biosimilares normalmente deben realizar ensayos clínicos, por lo que su desarrollo puede durar entre cinco y nueve años. No obstante, los medicamentos biosimilares representan un avance sustancial en la racionalización del gasto farmacéutico por su condición de medicamento similar a aquellos cuyas patentes han caducado. Hasta el momento, han reducido precios entre un 5 % y 35 % dependiendo de las moléculas o países 16.
Será muy relevante para el futuro la monitorización continua de la relación beneficio-riesgo en relación con la contención real del gasto sanitario, así como la experiencia obtenida a largo plazo, para poder realizar un adecuado análisis crítico del beneficio real de la introducción de los anticuerpos monoclonales biosimilares.
A modo de ejemplo, el ahorro estimado en el caso del Sistema Nacional de Salud de España es de 2443 millones de Euros para el periodo 2009-2020 (Figura 2).
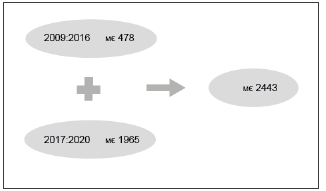
Fuente: Gonzalez A Yoana I, Zozaya N, Gimenez M, Hidalgo-Vega A. La Introducción de los biosimilares en España. Estimación del ahorro en el Sistema Nacional de Salud. Research Gate. DT N° 02/2017. Fundación Weber, Madrid, nov. 2017.
Figura 2 Ahorro estimado desde 2009 a 2020 como consecuencia de la introducción de los biosimilares en el SNS de España
Existe un amplio debate sobre los requerimientos a nivel de ensayos clínicos necesarios para la aprobación y registro de mAbs biosimilares. En torno a este tema, la agencia de registro de medicamentos europea (EMA) ha liderado el proceso y ha establecido criterios de bio-comparabilidad para el registro de estos productos. Así, en el 2013, los primeros mAbs biosimilares a Infliximab fueron comercializados en Europa, bajo los nombres de Resima e Inflectra.
Actualmente hay un fuerte interés en la introducción de mAbs biosimilares en diferentes mercados de Latinoamérica; en abril del 2015 Brasil aprobó el uso del primer AcMo biosimilar. La aparición de nuevas técnicas de producción, así como la introducción de los biosimilares anuncian una disminución en los precios de estos productos, lo que permitirá una mayor penetración de estos en mercados emergentes.
Temas como los criterios de calidad, seguridad y farmacovigilancia deben ser abordados y discutidos por las autoridades pertinentes con el fin de asegurar el beneficio de los pacientes 17.

Como evaluar un mAbs
El gran dilema de la evaluación económica en la sanidad radica en que las decisiones afectan la esperanza de vida de personas concretas afectadas por enfermedades graves. Una alternativa es evaluar, para cada unidad de salud producida, un año de vida ajustado por la calidad (AVAC), que equivale a un año de vida en perfecta salud. Este es el parámetro decisorio del National Institute for Clinical Excellence (NICE) británico, organización independiente responsable de proveer orientación para la promoción de la salud, la prevención y el tratamiento de las enfermedades en el Sistema Nacional de Salud en Inglaterra.
El NICE se apoya en los valores de coste por AVAC, cuando decide si recomienda o no un nuevo medicamento para su uso en el sistema de salud (NHS), aplicando un umbral de costo-efectividad de unas 30 000 libras esterlinas por AVAC ganado (NICE, 2008) 18. Un medicamento cuya relación coste/AVAC se sitúe por encima de esta ratio crítica tiene escasas posibilidades de ser incorporado a la práctica clínica en el sistema sanitario británico.
A este enfoque escapa el firme interés de cualquier enfermo de querer aplazar la vida por el hecho mismo, o en la espera de nuevas terapias. Queda claro que el AVAC no tiene en cuenta el criterio de equidad, que supera la óptica individualista, atendiendo a las necesidades sociales (paciente gravemente enfermo, estados de salud o de incapacidad crónicos). Además, la evidencia muestra que un año de vida ganado tiene mayor valor cuando tal ganancia se produce al final de la vida. Ante esta evidencia, NICE ha avanzado hacia un enfoque más social al considerar los tratamientos calificados como "end of life", aplicando una ponderación de 1,6 para los AVAC ganados. Este sería el caso de Sunitirib con una ratio de coste/AVAC de 50 mil libras. De todos modos, el criterio de AVAC es, hoy en día, un parámetro muy limitado y se debe seguir trabajando para mejorarlo.
En paralelo, la legislación sobre medicamentos en la Unión Europea establece el procedimiento centralizado, coordinado por la Agencia Europea de Medicamentos (EMA), como camino obligado para la evaluación y autorización de medicamentos biotecnológicos. La autorización de comercialización mediante el procedimiento centralizado es válida para todos los países de la UE y espacio económico europeo. Únicamente la fijación del precio del medicamento y su inclusión en el ámbito de aplicación de los sistemas nacionales de seguridad social, por motivos sanitarios, económicos y sociales, quedan a decisión de cada autoridad nacional 19.
DISCUSIÓN
Más allá del impacto en el diagnóstico de laboratorio, los anticuerpos monoclonales son una herramienta terapéutica poderosísima, entre otros factores por su alta especificidad y la posibilidad de facilitar distintos tipos de respuestas efectoras. Además, el empleo de anticuerpos humanizados y humanos ha mejorado notablemente su tolerancia clínica. La manipulación de los anticuerpos mediante la unión a otras moléculas o el diseño de nuevos fragmentos de anticuerpos abren un gran abanico de posibles aplicaciones en medicina.
El cáncer es uno de los grandes retos de la terapéutica actual. La investigación en profundidad de esta patología ha permitido facilitar su diagnóstico y desarrollar nuevas terapias. Gracias a estos avances, en los últimos años se ha reducido la mortalidad, y se ha mejorado significativamente la calidad de vida de los pacientes durante, y después del tratamiento.
El desarrollo de anticuerpos contra diferentes blancos tumorales, junto a los nuevos inhibidores farmacológicos, ha abierto la esperanza de poder visualizar el cáncer como un padecimiento crónico manejable y no como una enfermedad mortal. Si bien estos fármacos han demostrado su eficacia, son necesarios estudios de seguridad a largo plazo, así como evaluaciones de la relación coste-efectividad, comparando opciones terapéuticas equivalentes, para acabar de definir su papel en la práctica clínica.
Los retos actuales son desarrollar mAbs dirigidos hacia a nuevos blancos terapéuticos (para que se puedan administrar a pacientes que padezcan diferentes tipos de cáncer), mejorar el perfil de seguridad (evitar o reducir las reacciones adversas inmunes), conseguir un abaratamiento del coste de producción y distribución que en parte vendrá dado por el desarrollo de los biosimilares, que permitirán aplicar más eficientemente los fondos de los sistemas de salud con un mayor acceso, hasta abarcar a aquellos pacientes que hoy se encuentran privados de las nuevas terapias. Se ha de asegurar que el medicamento llegue en tiempo y forma, con la calidad y cantidad adecuada, y a un costo asequible para todos.
Esto se relaciona directamente con una posible revisión de la ley de protección de patentes. Como dato de referencia hemos de tener en cuenta que la Declaración Ministerial de Doha sobre el ADPIC y la salud pública es considerada por algunos autores como un avance sustancial en materia de acceso a los medicamentos. Esta declaración privilegia los intereses de la salud pública, por sobre los derechos de propiedad industrial, al separar los productos farmacéuticos de los demás productos comerciales: "reconocemos que la protección de la propiedad intelectual es importante para el desarrollo de nuevos medicamentos. Reconocemos asimismo las preocupaciones con respecto a sus efectos sobre los precios". A la vez, insiste sobre la importancia de la flexibilidad de las normativas sobe las patentes, destacando que "el Acuerdo sobre los ADPIC no impide ni deberá impedir que los Miembros adopten medidas para proteger la salud pública y, en particular, de promover el acceso a los medicamentos para todos".
De esta manera, se reconoce que existen situaciones en las que intereses públicos diferentes de los perseguidos por el sistema de patentes prevalecen sobre la defensa del derecho privado. Por esta razón, se debe procurar que esta declaración sea entendida como un logro esencial en cuanto a la protección de la salud pública y deba ser aplicado de manera que promueva el acceso a los medicamentos de forma universal *

