











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink1. INTRODUCCIÓN
Los compuestos con alta respuesta óptica no lineal (ONL) son de gran interés tecnológico debido a sus potenciales aplicaciones en el almacenamiento de datos, procesamiento de señales y tecnologías de telecomunicaciones [1,2]. Dentro de los compuestos que llaman la atención son los compuestos orgánicos de tipo “pushpull”, es decir, un compuesto con un fragmento donador de electrones (D) y otro aceptor de electrones (A) unidos covalentemente a través de un espaciador, el cual debe tener un sistema de electrones π deslocalizado.
Los benzotiazoles y sus derivados comprenden muchas aplicaciones, como en el campo biológico, farmacéutico, industrial debido a su papel como anticorrosivo de metales y como posibles materiales optoelectrónicos ya que la presencia de sistemas conjugados con deslocalización de los orbitales π, aumenta la probabilidad de que tengan lugar procesos de transferencia de carga. Además, es posible incorporar sustituyentes donadores y aceptores de carga, de tal forma, que le confieran una mayor transferencia [3-5]. Estas características hacen del 4-(6-(dimetilamino)benzo(d)tiazol2-il)benzonitrilo (figura 1) un candidato idóneo para aplicaciones de ONL [6,7].

Los métodos de la química computacional permiten evaluar las propiedades electrónicas de un sistema molecular y determinar si es un posible material con alta eficiencia opto-electrónicas, esto dependerá de muchos factores; como la fuerza donadora y aceptora de los sustituyentes, su posición en el sistema molecular, la asimetría del sistema y de la alta conjugación que se tenga, pero además por medio de esta herramienta se puede indagar sobre las propiedades espectroscópicas, estructurales y moleculares de los sistemas. Esto es un tema de sumo interés en el campo tecnológico, ambiental y particularmente en la investigación química debido a la cantidad de compuesto a los que se puede llegar por medio de la síntesis orgánica. En este trabajo se describió y caracterizó la estructura molecular, los orbitales naturales de enlace (NBO) y de frontera (HOMO, HOMO-1, LUMO y LUMO+1) y las propiedades de ONL, electronegatividad (x), dureza (n), suavidad (S), cargas NPA, el mapa de potencial electrostático (MEP) y los descriptores de reactividad global y local (funciones de Fukui) del 4-(6-(dimetilamino)benzo(d)tiazol2-il)benzonitrilo.
2. TEORÍA
El Mapa de Potencial Electrostático (MEP) es un método de cartografía del potencial electrostático, es decir, muestra la distribución de carga del sistema el cual se utiliza para determinar cómo las moléculas interactúan entre sí, así como también la forma, el tamaño y el momento dipolar de la molécula siendo así un método visual para comprender la polaridad relativa [8-10].
Los orbitales HOMO y LUMO son parámetros de energía que determinan la forma en que la molécula interacciona con otras especies. También se les conoce como orbital de frontera ya que el HOMO se define como el orbital más externo con mayor energía quien ocupa por lo menos un electrón, es decir es el último orbital ocupado electrónicamente y actúa como un donador de electrones ya que tiende a dar los electrones que en él se encuentren. Por otra parte, el LUMO se define como el orbital de menor energía más interno desocupado, es decir, el primer orbital desocupado electrónicamente quien puede aceptar electrones; [11] HOMO-1 y LUMO+1, representa el donante y aceptor, son niveles un estado de energía por debajo y por encima de estos niveles principales, respectivamente [12].
La hiperpolarizabilidad (β) la cual es una medida de la actividad óptica no lineal de un sistema molecular, está asociada con la actividad intermolecular de transferencia de carga resultante del movimiento de la nube de electrones a través de la estructura conjugada π, la cual va en dirección del grupo donante de electrones al grupo aceptor de electrones [13]. Las moléculas con un gran momento dipolar y un medio para cambiar la densidad electrónica tendrán hiperpolarizabilidades grandes, en general, se puede afirmar que dicha propiedad depende de la fuerza de los grupos dador y aceptor, así como de la longitud de la conjugación en el sistema que los une, por otra parte, en sistemas con anillos aromáticos sustituidos la plenaria afecta e influye en el tamaño del sistema de electrones π y en la movilidad de electrones. Se calcularon las propiedades ópticas no lineales del compuesto en estudio, como son el momento dipolar (µ), la polarizabilidad (α), y la hiperpolarizabilidad (β), las cuales usan las componentes x, y, z; siendo esta última la que determina la no linealidad óptica de las moléculas. Las ecuaciones (1), (2) y (3) [14,15] se emplearon para el cálculo de estos parámetros.
El momento dipolar estático total (µ), usando los componentes x, y y z se definen como:

La polarizabilidad promedio µtotal la cual se define como:
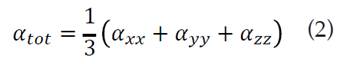
En la hiperpolarizabilidad de primer orden los componentes de β se definen como los coeficientes en la expansión en serie de Taylor de la energía en el campo eléctrico externo cuando el campo eléctrico externo es débil y homogéneo, esta expansión se da a continuación:

Por tanto:
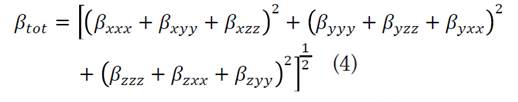
Propiedades como el potencial químico ( μ ) y la dureza ( η ) son expresadas sobre las bases de la aproximación de diferencias finitas en términos de I y A [10]. La suavidad ( S ) es un parámetro reciproco de la dureza, (x) corresponde a la electronegatividad, (w) equivale al índice de electrofilicidad; dichos parámetros se expresan en las siguientes ecuaciones:

La función de Fukui se interpreta como el cambio de potencial químico dada una perturbación externa o la variación de la densidad electrónica y se da al cambiar su número de electrones ya que cuando el sistema en estudio recibe o cede electrones se define la capacidad electrofílica o nucleofílica de la especie química. La función de Fukui refleja entonces la reactividad de diferentes sitios dentro de una molécula, es decir, la selectividad, en donde la dirección de ataque preferida por un reactivo será aquella donde la función de Fukui presente los valores más altos [16]. Por otro lado, la suavidad local se conoce como un indicador para las secuencias de reactividad intermolecular, dado que la suavidad local es una cantidad el cual es producto de un descriptor de reactividad global [16].
2. METODOLOGÍA
Se utilizó el software Gaussian09 revisión A02 [17] para llevar a cabo los cálculos químico-cuánticos y GaussView 5.08 [18] como visualizador. La optimización de la geometría se realizó al nivel de cálculo DFT/ cam-B3LYP/6-31G[2d) [19], se confirmó la ausencia de autovalores negativos de la matriz hessiana mediante un análisis vibracional. La frecuencia de las vibraciones del espectro IR teórico se corrigieron con un factor de escala de 0,9439 [20]. Con esta estructura se realizaron cálculos de energía para la molécula neutra e ionizada (positiva y negativa) y cargas del análisis de población natural (NPA) para determinar los centros nucleofílicos, electrofílicos y radicalarios, obteniendo posteriormente los descriptores de la reactividad local y las funciones de Fukui nucleófilica y electrofílica condensadas. A partir del cálculo TD-DFT se realizó el cálculo de estados excitados para obtener el espectro electrónico (UV-vis, emisión) y los orbitales de frontera HOMO-1, HOMO, LUMO y LUMO+1 para determinar la diferencia de energía (“Band-gap”). Las propiedades ópticas lineales y no lineales estáticas, momentos dipolares (µ), polarizabilidad (α) e hiperpolarizabilidad de primer orden (β), se obtuvieron mediante CP-DFT.
3. RESULTADOS Y DISCUSIÓN
3.1 Geometría molecular
La numeración de los átomos diferentes a hidrógeno usada en la estructura modelada se muestra en la figura 2.

Figure 2 Numbering of molecular structure of the 4-(6-(dimethylamino)benzo(d)thiazol-2yl)benzonitrile.
Los valores de las longitudes de enlace calculados se muestran en la figura 3. Los valores experimentales para los enlaces C2-S1 y C2-N3 son 1,80 y 1,32 Å [21] respectivamente; mostrando diferencias de 0,02 y 0,04 Å con respecto a los valores teóricos. Esto es consecuencia de que ambos heteroátomos están comprometidos con los electrones π de anillo benzoico lo cual acorta el enlace [22]. De igual forma, el enlace S1-C8 con un valor calculado de 1,73 Å, se acorta significativamente al ser comparado con el valor típico de 1,80 Å de un enlace sencillo C-S (figura 3). El valor experimental para los enlaces C=C del grupo fenilo es 1,40 Å, aproximadamente. Tomando este valor como referencia se puede observar en la figura 2 que las longitudes de enlace del fragmento fenilo están ligeramente afectadas (1,37 1,41 Å) [23]. Por otro lado, el grupo ciano (enlace C23-N24) con una longitud de enlace de 1,15 Å, muestra una variación respecto al valor experimental de 0,02 Å. El enlace C-N del sustituyente dimetilamino donde el nitrógeno cumple el papel de dador de electrones muestra una longitud de 1,37 Å, mostrando una excelente concordancia con el valor experimental (1,3740 Å) [24]. De acuerdo con los datos calculados y reportados experimentalmente, se puede concluir que existe una buena correlación en los valores de las longitudes de enlaces por lo que el nivel de cálculo DFT/camB3LYP/6-31G(2d). Cabe resaltar que, hasta donde se tiene conocimiento, no existe un reporte de las longitudes de enlace experimentales para el compuesto 4-(6-(dimetilamino)benzo(d)tiazol-2-il)benzonitrilo exactamente, por lo cual se tomaron fragmentos de su estructura para realizar el estudio comparativo.

Los ángulos del grupo fenilo del sistema benzotiazol oscilan entre 118° a 122°, que corresponde con el valor experimental de este tipo de ciclo con hibridación sp2 los cuales cuentan con un ángulo teórico de 120°. El ángulo de enlace que presentó un menor valor corresponde a los enlaces entre C2-S1-C8 con un valor de 89° y esto como consecuencia a la tensión anular que se genera en un anillo de cinco miembros. El sustituyente dimetilamino del anillo fenilo muestra un ángulo calculado de 119° causando una distorsión en la simetría del anillo, dando un valor menor de 120°, esto como consecuencia a su efecto electrónico.

Se pueden observar que en los enlaces C10-C11-C12-C13; C12-C13-C14-C15 se tienen ángulos calculados de 0°, al igual que en los enlaces C8-S1-C2-N3; S1-C2-N3-C9; C2-S1-C8-C9; C2-N3-C9-C8; C15-C10-C11-C12; C11-C12-C13-C14; C13-C14-C15- C10 tienen valores de -0,004°; -0,002°; 0,008°: 0,009°; 0,001°; -0,001° y 0,001° respectivamente, los cuales son valores muy cercanos a 0°; por otra parte entre los enlaces C8-S1-C2-C10; C10-C2-N3-C9; C4-C5-C6-N25; C2-C10-C11-C12; C23- C13-C14-C15 tienen valores de 179,980°; -179,986°; 179,522°; 179,995° y 179,999° respectivamente los cuales son valores aproximados a 180° lo que indica que el compuesto en estudio tiende a ser casi totalmente plano.
3.2 Análisis espectroscópico
3.3 Espectroscopia ultravioleta visible (UV-vis)
En la figura 5 se muestra el espectro ultravioleta en fase gaseosa, donde se obtuvo el máximo de absorción visible (λmax) y la fuerza del oscilador (f) para el único estado excitado de la molécula que ha sido reportado en la tabla 1.

Tabla 1 Energía, banda de absorción, fuerza del oscilador y transición de 4-(6-(dimetilamino)benzo(d)tiazol-2-il)benzonitrilo.
| Estado Excitado | Energía (eV) | Banda de absorción (nm) | Fuerza de oscilador | Transiciones |
|---|---|---|---|---|
| 1 | 2.5583 | 484.64 | 0.0000 | HOMO ( LUMO |
| 2 | 3.3148 | 374.04 | 0.0000 | HOMO-1 ( LUMO |
| 3 | 3.9130 | 316.85 | 1.1634 | HOMO ( LUMO |
| 4 | 3.9630 | 312.85 | 0.0000 | HOMO-1 ( LUMO |
| 5 | 4.0017 | 309.83 | 0.0000 | HOMO ( LUMO+3 |
| 6 | 4.2606 | 291.00 | 0.0000 | HOMO-3 ( LUMO+3 |
Se puede observar una banda de absorción a una longitud de onda de 331,19 nm y una fuerza de oscilador de 0,9115 atribuida a la transición permitida HOMO ( LUMO. Las 5 transiciones restantes no están permitidas (f = 0); mostrándose un espectro de longitud de onda larga que cubre la región entre 250 nm a 400 nm.
3.2.2 Espectroscopia infrarroja (IR)
En la mayoría de los espectros la zona que va de 2000 a 3000 cm-1 no presenta señales, solamente hay un grupo funcional que se identifica en esta zona, dicha señal corresponde a la presencia del grupo ciano (CN) cuyo valor experimental es de 2200 cm-1, en la tabla 2 se puede observar que esta señal es de 2271,98 cm-1, teniendo así buena correlación con el valor experimental y confirmando la presencia de este grupo funcional en el compuesto de estudio. Por lo general, las vibraciones de estiramiento de un sistema aromático C=C, dan lugar a una banda característica en los espectros IR cubriendo el rango espectral de 1400 a 1600 cm-1 [25]. En la tabla 2 se puede observar que esta asignación tiene un valor de 1609,65 cm-1 teniendo buena correlación con el rango experimental [26]. La vibración de estiramiento C=N del sistema benzotiazol se asigna a una banda experimental IR de intensidad muy fuerte a 1513 cm-1. En el espectro calculado se identifica esta banda debido a su intensidad, teniendo un valor calculado de 1519,05 cm-1 [27]. El estiramiento del C=C del sistema benzotiazol generalmente aparece en el rango de 1608 y 1584 cm-1, para el compuesto en estudio se asignó esta señal a 1306,58 cm-1 en el cual se da una deformación el anillo, dicho valor no se encuentra en el rango esperado; esto puede ser causado por el efecto de resonancia, o por la nube electrónica del sistema fenil [13]. Las vibraciones C-H en el plano son normalmente observadas en la región de 1300-1000 cm-1 como bandas débiles [17], en la tabla 2 se observa dichas vibraciones a 1168,85 cm-1 que involucran a los hidrógenos del sistema benzotiazol, del sistema fenil y del sustituyente dimetilamino. Por otra parte, esto nos confirma que el compuesto en estudio tiende a ser totalmente plano [14].

Tabla 2 Asignaciones de las señales de IR de 4-(6-(dimetilamino) benzo(d)tiazol-2-il)benzonitrilo.
| Experimental (cm -1 ) | Calculada (cm -1 ) | Escalada 0,9439 | Intensidad | Asignación |
|---|---|---|---|---|
| 1225 | 1222,17 | 1153,60 | baja | Flexiones C-H en el plano |
| 1430 | 1410,33 | 1331,21 | media | Estiramiento enlace C-N dimetilamino |
| 1384,24 | 1306,58 | media | Estiramiento C=C del benzotiazol | |
| 1513 | 1609,33 | 1519,05 | alta | Estiramiento C=N del benzotiazol |
| 1624 | 1705,32 | 1609,65 | baja | Estiramientos C=C |
| 2200 | 2407,02 | 2271,98 | baja | Estiramiento C-N del grupo CN |
3.3 Análisis HOMO-LUMO y descriptores de reactividad
Los valores calculados de energía de los orbitales frontera HOMO-1, HOMO, LUMO y LUMO+1 y “band-gap” se encuentran tabulados en la tabla 3. La figura 6 muestra que la mayor densidad electrónica de los orbitales HOMO se concentra en la región del grupo electrodonador, es decir, el dimetilamino, por tanto, se puede inferir que este sustituyente estabiliza la densidad electrónica de la molécula por el par de electrones libres que posee, dicha estabilización es consecuencia de los efectos de resonancia. Por otro lado, el orbital LUMO tiene la densidad electrónica distribuida sobre todo el sistema, pero en especial en la región del sustituyente electro-atrayente u aceptor el cual corresponde al grupo ciano, siendo así el orbital HOMO el dador y el orbital LUMO el aceptor. Por otra parte, la densidad electrónica del sistema espaciador se encuentra dispersa, no es notorio identificar que la densidad electrónica paso de una región a otra. En el HOMO -1 la densidad electrónica se centra en la parte derecha de todo el sistema, es decir, dicha densidad se encuentra concentrada sobre el sistema benzotiazol, por otra parte, el LUMO +1 la densidad electrónica se encuentra totalmente concentrada en el fragmento fenilo.
Tabla 3 Valores calculados para los orbitales de frontera y su respectivo “band-gap” de 4-(6-(dimetilamino)benzo(d)tiazol-2-il)benzonitrilo.
| Parámetro | Unidad (eV) |
|---|---|
| LUMO | 0,8289 |
| HOMO | -6,5234 |
| Band-gap | 7,3463 |
| LUMO+1 | 0,7671 |
| HOMO-1 | -7,8282 |
| Band-gap | 8,5359 |
Un “band-gap” de 7,3463 eV indica claramente que la molécula es muy estable, es decir, la transferencia de la carga está ocurriendo dentro de la molécula mediante el sistema espaciador. Sin embargo, el compuesto en estudio no sería un buen candidato para ser utilizado como semiconductor debido a que su valor de 7,3463 eV, ya que los compuestos más usados como semiconductores son el tiofeno; a través de la adición o eliminación de electrones de los orbitales π conjugados [8]. En general los materiales semiconductores orgánicos incluyen hidrocarburos aromáticos policíclicos (HAP) donde las propiedades conductoras dependen de la brecha energética y está establecido que la energía de banda de separación de los aislantes es grande (> 4 eV), pero inferior para semiconductores (< 3 eV) [9]. En el diagrama también se muestra el “band-gap” entre los orbitales HOMO-1 y LUMO + 1 de 8,5953 eV, lo cual para que se lleve a cabo una transición de electrones necesitaría mucha energía al tener este un valor mayor que el bang-gap de los orbitales HOMO-LUMO.
El potencial químico está relacionado con la electronegatividad, el cual indica que la densidad electrónica del sistema puede variar y los electrones pueden fluir de una región de alto potencial o mayor electronegatividad a uno de menor potencial o menor electronegatividad. El resultado obtenido para la molécula neutra (-3,6397 eV) nos indica que la densidad electrónica del sistema puede variar espontáneamente, al tener este un valor negativo [28]. Por otro lado, el valor de electronegatividad de 3,673 eV indica que la molécula tiende a atraer electrones sin cambiar su densidad electrónica. La dureza ( η ) corresponde a la separación entre el HOMO y LUMO, cuanto mayor es la brecha de energía orbital HOMO-LUMO, más dura es la molécula, lo que indica claramente que si la brecha de energía es mayor, la molécula será más dura, es decir mide la resistencia impuesta por éste al cambio en su distribución electrónica y/o polarizarse. Por tanto, el sistema al tener un valor de dureza de 2,850 eV tendrá poca tendencia a dar o recibir electrones, es decir, la dureza se ha asociado con la estabilidad del sistema químico. En ese orden de ideas y aplicando la teoría de ácidos y bases de Parr y Pearson [29,30], se tiene que el sistema en estudio posee un comportamiento de base dura, caracterizándose por su baja polarizabilidad, alta electronegatividad y por ser una especie donadora de electrones, por tanto, preferirá reaccionar con sistemas presenten el comportamiento de un ácido duro. La suavidad puede medir el grado de reactividad química del compuesto y ésta tiene un valor de 0,351eV y es el recíproco de la dureza [31].

3.4 Superficie de energía potencial (MEP)
En la figura 8 se ilustran los sitios reactivos de la molécula y la distribución de carga, de igual forma es muy útil para predecir sitios específicos en el cual se puede llevar a cabo un ataque electrofónico y/o nucleofílico.
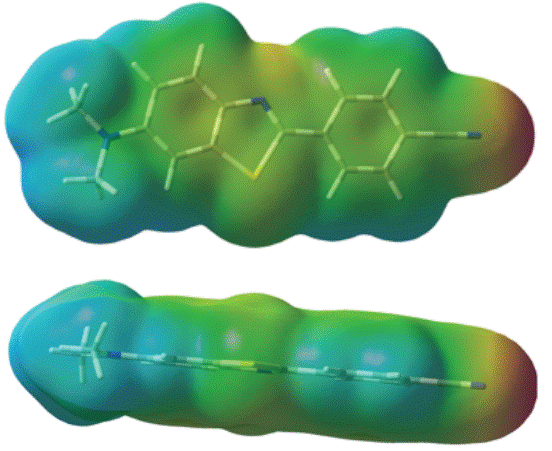
Figure 8 Map of electrostatic potential for the 4-(6-(dimethylamino) benzo(d)thiazol-2-yl)benzonitrile.
El rojo indica una alta densidad de electrones y el azul una concentración mínima de electrones, en la superficie interna de la molécula (regiones verdes), las densidades electrónicas son en promedio altos debido a la presencia de átomos de carbono que tienen menos electronegatividad, esto indica que el potencial electrostático es menor, teniendo así regiones azul oscuro indicadoras de extrema deficiencia electrónica, el cual se concentra en un extremo del compuesto en donde se encuentra el grupo dimetilamino N(CH3)2. En la parte intermedia del compuesto donde se encuentra el grupo fenil y benzotiazol presentan color verde lo cual indica que la distribución de electrones en esta zona es neutra o intermedia, es decir, que existe un balance de la densidad electrónica la cual se da por la presencia de enlaces covalentes y al otro extremo del compuesto donde se encuentra ubicado el grupo ciano (CN) hay una región que va desde un tono amarillo hasta un rojo intenso, dicha región indica que es rica en electrones. Por tanto, el grupo dimetilamino N(CH3)2 indica un posible sitio para el ataque nucleofílico ya que es un buen grupo saliente, por el contrario, en el grupo ciano (CN) se puede llevar a cabo un ataque electrofílico.
3.5 Descriptores locales de reactividad: funciones de Fukui y suavidad
Se realizó un análisis de las funciones condensadas de Fukui que se obtuvieron a partir del Análisis de Poblacional Natural (NPA), y así obtener las distribuciones de cargas para los estados aniónico (f -), catiónico (f +) y radicalario (f 0), que corresponden a la función de Fukui consensada, estos resultados se muestran en la tabla 5. Los valores de las funciones de Fukui negativos no se tendran en cuenta ya que no tienen significado físico puesto que los valores de estas funciones estan relacionado con la probabilidad de encontrar un electron en un orbital, lo que corresponde al cuadrado de la función de onda del sistema por tanto nunca se obtendrán valores negativos.
Observando los valores de f + en la tabla 5 se tiene que el valor más alto es de 0,1339 el cual corresponde al nitrógeno del grupo ciano (N24) debido a que se encuentra enlazado al carbono por un triple enlace provocándole características electrofílicas, el segundo valor más alto de f + corresponde al C con un valor de 0,1058; esto como consecuencia de ser el carbono donde se encuentra enlazado el grupo ciano (CN) el cual le atrae su densidad electrónica generando una desprotección convirtiendo a este carbono susceptible a un ataque nucleofílico. El tercer valor más alto de f + es 0,1055 para el N debido a que su par de electrones libres entra en resonancia con el sistema benzotiazol permitiendo que en un momento el nitrógeno tenga una carga parcialmente positiva, lo que se interpretaría como un centro electrofílico.
De igual forma, se puede analizar que el valor más alto de f - lo posee el átomo de nitrógeno del grupo dimetilamino (N25) con un valor de 0,5293 indicando que este es el átomo más susceptible para el ataque de un electrófilo como consecuencia al par electrónico solitarios que posee; siguiendo se tiene que el segundo valor más alto de f - es el C con un valor de 0,2777 debido a la alta densidad electrónica del sistema benzotiazol el fenil que hace parte de este anillo funcionado podrían experimentar reacciones de sustitución electrofílica aromática que conllevarían a activar la posición para. Uno de los valores más alto de f - corresponde a N con un valor de 0,1975, este nitrógeno había presentado propiedades electrofílicas, pero comparando su f+ y f- se encuentra que este último es mucho mayor, lo que indica que este nitrógeno tendrá mayor tendencia a reaccionar con electrófilos, ya que predomina la densidad del carga del sistema π sobre el efecto inductivo.
Tabla 5 Funciones de Fukui condensadas de 4-(6-(dimetilamino) benzo(d)tiazol-2-il)benzonitrilo.
| Átomo | f - | f + | f 0 | Átomo | f - | f + | f 0 |
|---|---|---|---|---|---|---|---|
| S1 | -0,1769 | 0,0893 | -0,0438 | H18 | -0,1091 | 0,0185 | -0,0452 |
| C2 | 0,0701 | 0,0280 | 0,0491 | H19 | -0,1230 | 0,0266 | -0,0481 |
| N3 | 0,1975 | 0,1055 | 0,1515 | H20 | -0,1327 | 0,0245 | -0,0541 |
| C4 | 0,0656 | 0,0432 | 0,0544 | H21 | -0,1204 | 0,0312 | -0,0446 |
| C5 | 0,2429 | 0,0130 | 0,1279 | H22 | -0,1204 | 0,0312 | -0,0445 |
| C6 | -0,0816 | 0,0547 | -0,0134 | C23 | -0,1554 | -0,0131 | -0,0842 |
| C7 | 0,2777 | -0,0006 | 0,1385 | N24 | 0,1937 | 0,1339 | 0,1638 |
| C8 | 0,0852 | 0,0170 | 0,0511 | N25 | 0,5293 | 0,0164 | 0,2728 |
| C9 | 0,1239 | -0,0316 | 0,0461 | C26 | 0,2101 | -0,0107 | 0,0997 |
| C10 | 0,0070 | 0,0733 | 0,0401 | H27 | -0,0793 | 0,0120 | -0,0336 |
| C11 | 0,1418 | 0,0412 | 0,0915 | H28 | -0,1040 | 0,0162 | -0,0438 |
| C12 | 0,0776 | 0,0274 | 0,0525 | H29 | -0,0850 | 0,0109 | -0,0370 |
| C13 | 0,1378 | 0,1058 | 0,1218 | C30 | 0,2101 | -0,0098 | 0,1001 |
| C14 | 0,0772 | 0,0285 | 0,0528 | H31 | -0,1036 | 0,0166 | -0,0435 |
| C15 | 0,1337 | 0,0426 | 0,0882 | H32 | -0,0793 | 0,0102 | -0,0345 |
| H16 | -0,1139 | 0,0170 | -0,0484 | H33 | -0,0856 | 0,0087 | -0,0384 |
| H17 | -0,1111 | 0,0213 | -0,0449 |
Debido a que el sistema en estudio es grande, las funciones de Fukui tienden a diluirse entre los átomos y en muchos de los casos su diferencia presenta valores pequeños. Para estos casos se hace uso de otros descriptores locales de reactividad; llamados suavidades locales los cuales complementan el estudio sobre los puntos más reactivos en la molécula. La suavidad local es directamente proporcional a la función de Fukui, por tanto en este caso los valores más altos en los tres parámetros (sf +, sf -, sf 0) lo tienen: sf + el átomo de carbono del sistema fenil (C13) con un valor de 0,5992, el átomo de nitrógeno del grupo dimetilamino (N25) tiene el mayor valor de sf -, sf 0 con 2,5977 y 1,3391 respectivamente, siendo estos átomos los que permiten un mayor cambio en su densidad electrónica al interaccionar con un electrófilo, nucleófilo o radicales.
3.6 Orbital natural de enlace (NBO)
El análisis de NBO proporciona un método eficiente para estudiar la interacción intra e intermolecular [32], también es una herramienta esencial para investigar la transferencia de carga o conjugación en sistemas moleculares [33], en términos de ocupación y composición de los NBOs Lewis. Se pudo observar que las densidades electrónicas u ocupaciones máximas son 1,99256; 1,99043; 1,99736; 1,99439; 1,99193; 1,99435; 1,99436 que corresponde a los enlaces tipo sigma (σ) de C2-N3; C13C23; C23-N24; C23-N24; C6-N25; N25-C26 y N25-C30 respectivamente, mostrando una fuerte interacción de los electrones tipo sigma. Estas altas interacciones y densidad electrónica se encuentra en todo el sistema, ya que se obtienen valores altos a un extremo del sistema que corresponde a la ubicaciones grupo ciano que se encuentra en posición para con respecto al fenilo (CN) (C13-C23; C23-N24); en el centro de la molécula (C2-C10) y en el otro extremo de la molécula donde se encuentra ubicado el grupo dimetilamino (C6-N25; N25-C26; N25-C30); este último puede ser un factor determinante en los valores más altos de densidad electrónica, debido a que el nitrógeno en sus enlaces tiende a atraer cierta densidad. En los orbitales tipo Lewis, el enlace σ(S1-C2 ) se forma a partir del híbrido sp4.89 sobre el azufre, que es la mezcla de s (16,91%) p (82,63%) d (0,47%); de igual forma el enlace σ(C -N ) se forma a partir del híbrido sp1.89 sobre el carbono, que es la mezcla de s (34,59%) p (65,32%) d (0,09%); por otra parte el enlace π (C2-N3) se forma a partir del híbrido sp sobre el carbono y el nitrógeno, que es la mezcla de s (0,00%) p (99,88%) d (0,12%), lo que da lugar a la formación de un enlace doble remarcado por el orbital p. Los pares electrónicos solitarios estabilizan el sistema generando una transferencia de carga intramolecular al tener valores muy cercanos a las contribuciones para la formación de un enlace sigma (σ) y para un enlace pi (π) como por ejemplo 1,99062; 1,94395; 1,98500 del LP(1) S1; LP(1) N3 y LP(1) N25; respectivamente, sin embargo estos pares electrónicos libres cuentan con diferente hibridación ya que los pares solitarios S1 LP(2) y el N25 LP(1) poseen hibridación p1.00, puesto que un par electrónico libre es un orbital p puro; pero el N3 LP(1) posee hibridación sp2.45 es decir, la hibridación de este par electrónico libre corresponde a sp2 ya que este hace parte del anillo aromático del sistema benzotiazol.
3.7 Propiedades de óptica no lineal (ONL)
Las componentes vectoriales del momento dipolar calculado son μx = 8,2642 D, μy = -0,3694 D y μz = 0,2988 D, y el módulo es 8,2778 D. La componente μx tiene la mayor contribución, debido a que ese es el eje que contiene los grupos donde tiene lugar la transferencia de carga, del grupo dimetilamino hacia el grupo ciano. La polarizabilidad del sistema de electrones π se logra uniendo grupos electro donadores y electro aceptores al principio y al final de los anillos o sistemas de anillos, con esto se crea un eje de transferencia de carga, por tanto, la polarizabilidad de una molécula es la medida de la capacidad de responder a un campo eléctrico y adquirir un momento eléctrico dipolar.
Tabla 6 Momento dipolar (μ), polarizabilidad (α) e hiperpolarizabilidad (β) de 4-(6-(dimetilamino)benzo(d)tiazol-2-il)benzonitrilo.
| Parámetro | Debyes | Parámetro | esu | Parámetro | esu |
|---|---|---|---|---|---|
| µtotal | 8,2778 | αtotal | 3,528x10-23 | (βtot) | 5,3509x10-29 |
| ∆α | 6,469x10-23 |
Los valores obtenidos para las polarizabilidades promedios se muestran en la tabla 6, al igual que, el valor de la anisotropía de la polarizabilidad ∆α (dependencia de la orientación molecular respecto a un campo eléctrico aplicado) en función de sus tensores (fuerza ejercida sobre una superficie de la molécula) sobre los ejes x, y, z. Se puede observar que el valor de polarizabilidad total (αtotal) da como resultado un valor positivo (3,528 x 10-23 esu) lo que indicaría que no existe perdida de linealidad óptica del sistema molecular, consecuencia tanto de la asimetría del compuesto como el alargamiento del sistema conjugado. La anisotropía calculada de la polarizabilidad del compuesto en estudio es de 6,469 x 10-23 esu, lo que quiere decir que cuando un campo eléctrico paralelo o perpendicular al eje molecular incide sobre la molécula distorsionando así su nube electrónica con facilidad por la presencia de un ion cercano o un dipolo. Al tener un valor de hiperpolarizabilidad (β) de 5,3509 x 10-29 esu el compuesto sería un material con alta respuesta óptica no lineal, esto debido a la deslocalización de electrones a lo largo de un esqueleto conjugado y a la asimetría que se presenta gracias a los dos grupos, uno aceptor como el grupo ciano (CN) que entraría en resonancia con el sistema aumentando así la hiperpolarizabilidad y un grupo altamente electrodonador como lo es el grupo dimetilamino (N(CH3)2) que hace una mayor contribución al activar el anillo benzotiazol aumentando su densidad electrónica hacia el grupo ciano quien se encuentra en posición para con respecto al fenil creando un eje de carga preferencial aumentando sus características de ópticas, pero además la electronegatividad y el par de electrones solitarios de los átomos de azufre (S) y del nitrógeno (N) del sistema benzotiazol junto con la extensión de la longitud de conjugación del puente π-electrón también conduce a un aumento de la hiperpolarizabilidad molecular, por tanto, se puede considerar al compuesto en estudio como un posible candidato para estudios experimentales de óptica no lineal. El valor de βtotal obtenido para el compuesto en estudio ha sido comparado con el de la urea, el cual es el compuesto de referencia que tiene un valor de β total = 0,0654 x 10-29 esu. Como se muestra en la tabla 6 al tener un valor de β = 5,351 x 10-29 esu el compuesto en estudio y β = 0,0624 x 10-29 esu para la urea al mismo nivel de cálculo se tiene una relación de 81,82 lo que afirmaría que el compuesto puede ser un óptimo candidato para estudios experimentales de óptica no lineal y posteriormente para aplicaciones optoelectrónicas.
4. CONCLUSIONES
Por medio del nivel de cálculo DFT/cam-B3LYP/631G(2d) se realizó la optimización la geometría del compuesto para establecer las longitudes de enlaces, ángulos de enlace y los ángulos diedros de la molécula y por medio de esto se logró concluir la planaridad que posee y mediante la optimización se obtuvieron datos teóricos acerca de las transiciones en el UV-Visible y de espectroscopia infrarroja (IR). Se lograron determinar la energía de los orbitales HOMO-LUMO con su respectivo band-gap, presentando estas buenas propiedades electrónicas, se calculó y analizó el mapa de potencial electrostático (MPE) teórico y se logró establecer los sitios de mayor y menor densidad electrónica, por medio del análisis NBO se obtuvo la hibridación de cada orbital molecular, su ocupación electrónica y la contribución respectiva de los orbitales para los átomos más representativos, usando las funciones de Fukui se pudo establecer los sitios más susceptibles para que se lleve a cabo reacciones nucleofílicas, electrofílicas y radicalarias y por último, el compuesto en estudio presento una alta relación de óptica no lineal con respecto a la urea, por lo que puede considerarse un posible candidato para aplicaciones de materiales ópticos y electrónicos.

