











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
Introducción
El colágeno es una estructura proteica que se encuentra en todos los órganos del cuerpo humano. Existen diferentes tipos de colágeno, el tipo IV es el componente principal de la membrana basal glomerular (MBG) madura, cóclea y ojos. El colágeno tipo IV en la MBG está conformado por tres cadenas (α1, α2, α3) que se entrelazan entre sí, formando un enrejado. Mutaciones en los genes COL4A3, COL4A4 y COL4A5 afectan la síntesis, ensamblaje y función del colágeno IV α345, ocasionando una nefropatía hereditaria denominada síndrome de Alport (SA), que se puede manifestar clínicamente desde hematuria microscópica hasta enfermedad renal crónica (ERC) (1).
Históricamente, la glomerulonefritis membranoproliferativa fue clasificada en tres grandes tipos, dependiendo de la presencia y la ubicación de los depósitos electrodensos vistos en la microscopia electrónica: subendoteliales, subepiteliales e intramembranoso. Pero ahora se han reconocido casos que muestran solo depósitos del componente C3 de la vía del complemento, debido a un control anormal de la vía alternativa del complemento se denominan glomerulopatías C3 e incluye: la enfermedad de depósitos densos (EDD), la cual muestra depósitos de características muy densas en la MBG; y la glomerulonefritis C3 (GNC3) es definida como la presencia de forma aislada o dominante de depósitos de C3 en la inmunofluorescenica, ubicados en distintos sitios del glomérulo y no solo en la MBG como en la EDD (2).
A continuación se reporta el caso de un niño escolar con SA ligado al cromosoma X, que desarrolla una glomerulonefritis y presenta manifestaciones clínicas mixtas para síndrome nefrítico y nefrótico.
Caso clínico
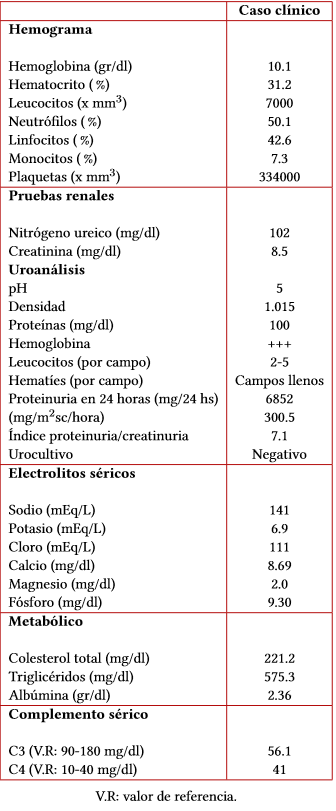
Paciente masculino de once años de edad quien fue remitido de una institución del área rural por cuadro clínico de cinco días de evolución consistente en oliguria, hematuria, edema facial y en miembros inferiores; había presentado faringoamigdalitis quince días previos al inicio de la sintomatología. Los paraclínicos realizados en el centro de atención inicial mostraron creatinina sérica de 7.3 mg/dl BUN: 88 mg/dl, parcial de orina con sangre +++ proteínas: 300mg/dl, por lo que remiten. Al examen físico de ingreso se encuentra: peso 26.8 kg (z:-2,28), talla 128 cm (z:-2,80), tensión arterial 90/50 mm Hg, edema facial, ascitis y edema de miembros inferiores. Sin antecedentes patológicos conocidos. En la Tabla 1 se muestran los paraclínicos realizados a su ingreso, en los que se documentó elevación de azoados, hematuria, proteinuria en rango nefrótico, dislipidemia, hipoalbuminemia y C3 disminuido. Se realiza además una ecografía renal que evidencia aumento difuso de ecogenicidad del parénquima renal. Ingresa a unidad de cuidados intensivos (UCI) por urgencia dialítica e inicia diálisis peritoneal. Durante su estancia en UCI presenta emergencia hipertensiva con órgano blanco cerebro, por lo que se inicia además terapia antihipertensiva. Por sospecha de glomerulonefritis rápidamente progresiva se colocan tres pulsos de Metilprednisolona y dosis mensual de Ciclofosfamida (cuatro en total). Se realiza biopsia renal la cual reporta en la microscopía óptica proliferación celular endocapilar/mesangial difusa; la inmunofluorescencia mostró depósitos de C3 (figura 1), y la microscopía electrónica muestra presencia de depósitos electrodensos subepiteliales, subendoteliales e intramembranosos en vías de reabsorción, con lo que se realiza el diagnóstico de glomerulonefritis C3 y se sospecha una mutación de la vía alterna del complemento, por lo que se realiza estudio genético del complemento, el cual mostró una deleción heterocigota en el Gen CFHR1 que compromete los exones 4-6, variante de significado incierto, mostrado en la figura 2. Es importante aclarar que no se pudo realizar anticuerpos con factores del complemento (Anti-CFH) ni determinación de C3Nef, por trámites administrativos.

Fuente: elaboración propia.
Figura 1 Estudio histopatológico: A) microscopia óptica: proliferación celular endocapilar y mesangial difusas, membrana basal con dobles contornos. B) inmunofluorescencia: presencia dominante de C3 en mesangio y en membrana basal glomerular.

Fuente: documento obtenido de la historia clínica.
Figura 2 Pruebas moleculares. A. Prueba genética molecular del complemento. B. Prueba genética molecular de Síndrome nefrótico corticorresistente.
El paciente requirió terapia de reemplazo renal durante quince días. Luego presentó mejoría parcial inicial de edema en cara y miembros inferiores, con mejoría de los trastornos electrolíticos y de la función renal, pero con persistencia de proteinuria en rango nefrótico. Por respuesta inadecuada a las ocho semanas del tratamiento inmunosupresor con esteroides se declara corticorresistente; se inicia manejo con Tacrolimus y se realiza el panel genético para síndrome nefrótico corticorresistente y descartar mutaciones genéticas de enfermedades glomerulares, sorpresivamente mostró la variante COL4A5 (ver reporte en la figura 2), asociada al síndrome de Alport ligado al X. Nunca se pensó en esta entidad al no tener antecedentes familiares de sordera. Hasta la fecha el paciente no ha presentado alteraciones en la evaluación auditiva, mientras que en la evaluación oftalmológica se evidenciaron cataratas bilaterales asintomáticas. Actualmente, el paciente se encuentra en tratamiento interdisciplinario, recibiendo prednisolona y Tacrolimus. Por compromiso acelerado de función renal se retira Tacrolimus y se inicia micofenolato de mofetilo. En la Tabla 2 se resumen los principales aspectos clínicos, paraclínicos y de tratamiento en la evolución del paciente, donde se puede observar la progresión de la ERC y la persistencia de la proteinuria en rango nefrótico. A la fecha actual, el paciente presenta compromiso de función renal con disminución progresiva de tasa de filtración glomerular a 20 ml/min/1,73. Por presentar la asociación de dos entidades fisiopatológicas distintas y poca probabilidad de recuperación de la función renal deteriorada es derivado a cita de pretrasplante renal, con la finalidad de poder ofrecer un trasplante preemptive (anticipado).

Discusión
El SA es una nefritis hereditaria que histológicamente se caracteriza por un engrosamiento y lamelación de la MBG; en la microscopia electrónica se pueden encontrar depósitos electrodensos pero generalmente con inmunofluorescencia negativa (3,4). Se produce principalmente por una mutación en los genes que codifican el colágeno tipo IV (COL4). El tipo ligado al cromosoma X (en el 85 %) es debido a una mutación en la cadena α5 del COL4 (COL4A5). Existen casos más raros que se heredan de forma autosómica recesiva o autosómica dominante por mutación en la cadena α3 del COL4 (COL4A3) y en la cadena α4 del COL4 (COL4A5) (3,5). La prevalencia del SA es 1 en 50.000 nacidos vivos (6). El curso clínico es muy variable, puede presentar hematuria micro y macroscópica, proteinuria, hipertensión, sordera y algunos pueden progresar a enfermedad renal crónica terminal (7). Además se han descrito múltiples manifestaciones oculares las cuales pueden ser asintomáticas (8) como las cataratas que presenta este paciente. En muy raras ocasiones produce síndrome nefrótico como en el caso reportado (9).
La GNC3 es una entidad que generalmente presenta un patrón de glomerulonefritis membranoproliferativa en la microscopía de luz, con depósitos de C3 en el estudio de inmunofluorescencia y en la microscopia electrónica se pueden encontrar depósitos electrodensos mesangiales, subendoteliales, subepitelilaes o intramembranosos como en el caso clínico de este reporte (10-12). La GNC3 se produce debido a una hiperactividad de la vía alternativa del complemento por defectos en las proteínas reguladoras de esta vía del complemento, lo que genera depósitos en la pared de los capilares glomerulares (13). Diferentes estudios han encontrado mutaciones en genes del complemento: CFH (del inglés, factor complement H), CFI (del inglés, factor complement I), MCP (del inglés, membrane-cofactor protein), CFB (del inglés, factor complement B), C3, CFHR (proteínas relacionadas con CFH) y autoanticuerpos contra el CFH, CFB y C3 convertasa (conocido como factor C3 nefrítico, C3Nef) (11). Su presentación clínica es muy variada, desde síntomas leves como microhematuria o proteinuria, hasta síntomas severos como síndrome nefrítico, nefrótico y daño renal (11).
El paciente reportado cursó con manifestaciones clínicas de un síndrome nefrótico corticorresistente; se le practicó una biopsia renal que mostró hallazgos de una glomerulonefritis mebranoproliferativa, con presencia de C3 en mesangio y membrana basal; en la microscopía electrónica se evidenciaron depósitos electrodensos subendoteliales, subepiteliales e intramembranosos en vías de reabsorción. Se le realizó test genético para las proteínas reguladoras de la vía alternativa del complemento y se encontró una deleción heterocigota en el gen CFHR1 (proteína 1 relacionada al factor H). Al no presentar respuesta a la terapia inmunosupresora con esteroides durante ocho semanas se declara corticorresistente y se le realiza test genético para síndrome nefrótico corticorresistente, sorpresivamente se encuentra una mutación en el gen COL4A5 asociada a SA ligada al X. Recibió tratamiento con Micofenolato de Mofetilo asociado a esteroides durante dos años, siendo el ahorrador de esteroides que mejores resultados ha demostrado en GNC3 (14,15). Una opción terapéutica para el paciente reportado podría ser el bloqueo de la ruta alternativa del complemento con Eculizumab. Existen estudios que muestran que un grupo de pacientes con GNC3, no todos, podrían obtener beneficio clínico con esta terapia (16).
La presentación de síndrome nefrótico es muy rara en el SA (17), pero si es frecuente que se presente en la GNC3 (11,12). El paciente reportado cursó con manifestaciones clínicas de síndrome nefrótico y síndrome nefrítico (proteinuria nefrótica, hipoalbuminemia, dislipidemia, edemas, hematuria, hipocomplementemia, hipertensión, daño renal agudo) que se explican por la presencia de glomerulonefritis aguda. A pesar que el 80 % de los hombres con SA desarrollan hipoacusia neurosensorial y hasta el 40 % presentan problemas oculares (18,19), en el seguimiento de este caso clínico no se ha encontrado hipoacusia.
Desde décadas pasadas se han reportado casos de SA imitando glomerulonefritis mediada por complejos inmunes (20-23), Nars SH y colaboradores muestran cinco casos de nefritis hereditaria con depósitos electrodensos en la microscopia electrónica, con inmunofluorescencia positiva en cuatro casos, sugiriendo glomerulonefritis mediada por complejos inmunes, aunque no hubo evidencia de glomerulonefritis activa (23), ya que no se evidenció proliferación endocapilar como la presentada en nuestro paciente. Además, en la literatura actual se encuentran reportes de algunos casos de SA asociados a glomeruloesclerosis focal y segmentaria (24), nefropatía membranosa (9), síndrome hemolítico urémico (25) y vasculitis (26). El caso reportado muestra la asociación de SA con una GNC3, con depósitos en mesangio y membrana basal e IF positiva solamente para C3. Se descartó la presencia de enfermedades autoinmunes y el diagnóstico de nefritis hereditaria fue posteriormente realizado en el test genético.
Conclusión
Tanto el SA y la GNC3 son entidades raras, que excepcionalmente pueden encontrarse asociadas, pero es importante que el clínico tenga conocimiento que pueden existir casos en que un trastorno puede estar asociado a otro, debido a que poseen bases fisiopatológicas completamente distintas. Se presentó un paciente pediátrico que cursó con dos entidades clínicas que afectan al riñón, pero con diferentes mecanismos fisiopatológicos: (i) la alteración estructural de la membrana basal del glomérulo, producida por una mutación en las cadenas del colágeno IV como es el SA, y (ii) la desregulación de la vía alternativa del complemento conlleva a la producción de depósitos en las paredes de los capilares. Es importante resaltar que el síndrome de Alport puede manifestarse como un síndrome nefrótico corticorresistente y presentarse concomitante con otras enfermedades glomerulares, en este caso GNC3. Estas condiciones, al estar asociadas, podrían acelerar la progresión a enfermedad renal crónica terminal. Por lo anterior, se hace necesario que los pacientes que presenten estas asociaciones se les realicen un seguimiento clínico más estricto para ofrecer un tratamiento temprano y oportuno.

