











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkINTRODUCCIÓN
La esclerosis sistémica (ES) es una enfermedad autoinmune, crónica, discapacitante, caracterizada por tres aspectos fundamentales: el compromiso microvascular (proliferativo y obliterativo), la activación del sistema inmune y el aumento de los depósitos de matriz extracelular en la piel y los órganos internos1.
Hasta hace 35 años la crisis renal era la principal causa de mortalidad, pero esto cambió radicalmente con la introducción de los inhibidores de la enzima convertidora de angiotensina (iECA). Por este motivo, la enfermedad pulmonar con sus dos manifestaciones: hipertensión pulmonar (HTP) y enfermedad pulmonar intersticial difusa (EPID), pasó a ser la principal causa de mortalidad, dando cuenta del 30 y 35% respectivamente2. Esto se evidenció en la cohorte de Pittsburgh, Seguimiento a largo plazo, y se vio un cambio en la mortalidad por crisis renal que pasó de 42 % a 6 %; al contrario, la enfermedad pulmonar aumentó de 6 a 33%3, convirtiéndose en una prioridad en el tratamiento de los pacientes con ES.
MÉTODOS
Se hizo una revisión narrativa de la literatura en Pubmed desde su creación hasta la fecha en inglés y español utilizando los siguientes términos MeSH: enfermedad pulmonar intersticial, esclerosis sistémica, tomografía, ciclofosfamida, micofenolato mofetil, azatioprina, rituximab, trasplante de médula ósea. No se limitó la búsqueda por fecha. Se incluyeron 52 artículos, después de excluir aquellos en que coexistían EPID e HTP así como los que se refirieran a EPID en el contexto de enfermedad del tejido conectivo no limitada a ES.
EPIDEMIOLOGÍA
La enfermedad pulmonar intersticial (EPI) es una afección común en la ES. Su frecuencia es difícil de definir y varía según la serie evaluada4)(5)(6, dado que depende del parámetro usado para diagnosticarla: tomografía computarizada de alta resolución (TCAR), pruebas de función pulmonar o, incluso, reporte de necropsia. Según la serie de la Liga Europea contra el Reumatismo (EULAR) de 20076, en la variedad difusa 53,4 % tenían fibrosis pulmonar; 49,3 %, compromiso restrictivo y 64 %, alteración de la difusión de monóxido de carbono (DLCO); en cambio, en la variedad limitada 34,7 % tenían fibrosis pulmonar; 26,7 %, defecto restrictivo y 71,8 %, disminución de la DLCO. Este último valor tan alto se explica porque la DLCO también sirve como prueba de tamización en hipertensión pulmonar, que se presenta más en los pacientes con la variedad limitada y por tanto no debe tomarse como un indicador absoluto de EPID; la DLCO no es un marcador diagnóstico de enfermedades respiratorias específicas, sino un reflejo fisiológico del intercambio gaseoso alveolar, que se puede afectar por alteraciones parenquimatosas y vasculares pulmonares y se utiliza más con fines pronósticos; también se debe reconocer su variabilidad entre centros y la importante influencia, en su resultado, de la colaboración del paciente durante su ejecución. Con respecto a la historia natural de la enfermedad, 50 % de los pacientes la desarrollan en los primeros tres años y el riesgo disminuye posteriormente7. Alrededor de 16 % tienen compromiso grave que se define como una capacidad vital forzada (CVF) menor de 55 %, lo que disminuye la supervivencia a nueve años a solo 38 %7.
FISIOPATOLOGÍA
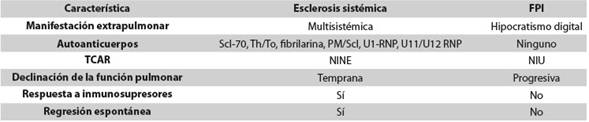
Aún no hay claridad sobre muchos aspectos de la patogénesis de la enfermedad pulmonar; se ha propuesto que hay una interacción entre la inmunidad innata y la adquirida que genera inflamación y fibrosis. El daño endotelial y epitelial recurrente promueve el reclutamiento de macrófagos y linfocitos, lo que resulta en la producción de mediadores profibróticos incluyendo el factor de crecimiento transformante β1, el factor de crecimiento del tejido conectivo y el factor de crecimiento derivado de las plaquetas, que llevan a la activación, proliferación y supervivencia de fibroblastos, y a la diferenciación al fenotipo de miofibroblastos contráctiles, con la consecuencia de sobreproducción y acumulación de la matriz extracelular8. En el lavado broncoalveolar hay aumento de los eosinófilos y en la sangre periférica, de la expresión de CD48, el cual puede actuar como molécula de adhesión y atraer eosinófilos a tejidos periféricos como el pulmón, aunque no existe una relación directa entre la magnitud de expresión y el daño9. En ciertos aspectos, esta patogénesis es similar a la de la fibrosis pulmonar idiopática (FPI); se resaltan, como aspectos comunes, los siguientes eventos que ocurren en el epitelio alveolar y en el endotelio vascular: apoptosis, autofagia, broncoaspiración, activación de la cascada de la coagulación y de la inmunidad innata y adquirida, aumento de citocinas y quimiocinas y activación de los fibroblastos; sin embargo, el comportamiento de los pacientes con ES es completamente distinto y susceptible a inmunosupresión (Tabla 1)10)(11)(12.
MANIFESTACIONES CLÍNICAS
El síntoma más común es la disnea con el ejercicio, otros son: tos, fatiga y dolor torácico. Al examen físico se suelen encontrar crépitos en velcro predominantemente en las bases; no obstante, un gran porcentaje de pacientes permanecen asintomáticos y el compromiso pulmonar ya está avanzado cuando se evidencian los hallazgos en el examen físico, lo que afecta negativamente la morbimortalidad13.
También es importante mencionar que el compromiso pulmonar intersticial puede ocurrir en la ES sin afección cutánea; este fenómeno se ha denominado esclerosis sistémica sine esclerodermia; en las diferentes cohortes, su prevalencia oscila alrededor de 8,3 %; en estos pacientes, el compromiso pulmonar es el segundo más frecuente, luego del esofágico.
DIAGNÓSTICO
Dado que muchos pacientes son asintomáticos, las pruebas de función pulmonar tienen un papel fundamental. El compromiso es variable dependiendo del método que se utilice para su detección, así: hasta 40 % - 75 % de los pacientes tienen cambios restrictivos en la espirometría; 90 %, alteraciones en la TCAR14 y 100 %, hallazgos de EPID en la necropsia15. El hallazgo característico en la espirometría es un patrón restrictivo, con disminución de la capacidad vital forzada (CVF), pero son normales el volumen espiratorio en el primer segundo (VEF1) y la relación VEF1/CVF; la difusión de monóxido de carbono (DLCO), por su parte, se encuentra disminuida, dado que hay un engrosamiento del intersticio que disminuye el intercambio; esto suele ocurrir muy tempranamente16. En estudios recientes se ha visto que no solo la DLCO en reposo puede ser predictora de EPI, sino también la llevada a cabo con un esfuerzo físico, que puede estar alterada incluso antes que la de reposo17. Otra de las pruebas que se pueden hacer es la caminata de seis minutos, que lleva a un ejercicio aeróbico submáximo que se correlaciona con la actividad física diaria; podría ser útil cuando sea difícil hacer la espirometría; sin embargo, es controversial su utilidad para el diagnóstico de EPI en ES, dado que puede no detectar hasta 30 % de los pacientes con enfermedad18. Se utiliza más para evaluar el desempeño cardiopulmonar, afectado de manera importante en los pacientes con ES por su desacondicionamiento físico, el fenómeno de Raynaud y la miopatía asociada y tiene mayor utilidad como desenlace objetivo en las terapias para hipertensión arterial pulmonar.
Un estudio reciente mostró que una desaturación mayor de 4 % durante la caminata de seis minutos tiene una correlación de 0,78 con una DLCO baja19. Sin embargo, la evidencia es heterogénea por lo que los metaanálisis no han dado resultados concluyentes, especialmente por la variabilidad en el protocolo seguido para la prueba20.
Con respecto a las imágenes diagnósticas, la radiografía de tórax carece de sensibilidad para el diagnóstico de EPI, por cuya razón la TCAR es la técnica ideal21)(22; en ella puede haber diversos hallazgos como patrón de vidrio esmerilado, panal de abejas de distribución basal y periférica y engrosamientos pleurales; todos ellos se pueden ver tempranamente y tienen una correlación alta con la histología por lo que raramente es necesaria la biopsia pulmonar12. Habitualmente no se hace la puntuación formal, pero un cálculo rápido semicuantitativo de la extensión de la enfermedad en la TCAR que demuestre compromiso de más del 20 % del parénquima pulmonar, combinado con un umbral de la CVF menor de 70 % se ha utilizado para calificar la etapa de la enfermedad como limitada o extensa lo que define a los pacientes con peor pronóstico y, por tanto, a aquellos que se benefician de la terapia inmunosupresora23. En la actualidad existen varias herramientas informáticas para segmentar automáticamente el pulmón, que le permiten al clínico definir con facilidad el porcentaje de afectación; ellas son: visualización de la imagen, cuantificación anatómica y caracterización regional del tejido pulmonar24. Se ha visto que el engrosamiento pleural es muy característico en la EPID por ES, por lo que la ecografía ha surgido como alternativa para el diagnóstico temprano, con la ventaja de disminuir la exposición a la radiación; en los diferentes estudios se ha encontrado que un engrosamiento pleural mayor de 2,8-3 mm en varias zonas pulmonares es muy sugestivo de ES como etiología de la EPID. Otros hallazgos como las líneas B en el parénquima pulmonar, aunque presentes en un alto porcentaje de pacientes con compromiso intersticial por ES, no son específicos de la enfermedad25)(26; son pocos los estudios y falta aún validación de este método, sin olvidar que depende mucho del operador (Tabla 2).

EVALUACIÓN DE LA PROGRESIÓN
La progresión ha sido difícil de predecir, por lo que se recomienda el seguimiento anual con TCAR, espirometría y DLCO; se consideran como cambios significativos la disminución del 10 % en la CVF o del 15 % en la DLCO o un compromiso mayor del 20 % en la TCAR2. Además, en algunos estudios clínicos se han encontrado variables que pueden llevar a la progresión o a la EPI; ellas son:
El sexo y la raza: la ES suele afectar más a las mujeres; cuando se presenta en hombres es más grave y más aún si son afroamericanos, quienes también hacen más tempranamente compromiso pulmonar. Este último hecho se puede explicar por la diferente expresión de citocinas en el lavado broncoalveolar de los pacientes afroamericanos, quienes expresan más TGF-β1, entre otras citocinas; esto no solo se evidencia en pacientes con ES, sino también en población sana, lo que podría sugerir que el paciente afroamericano tiene un ambiente de citocinas profibrótico que lo predispone a desarrollar EPI intersticial y a tener un peor pronóstico27.
Extensión del compromiso de la piel: en la cohorte EUSTAR (EULAR Scleroderma Trials And Research) se encontró que los individuos con ES de la variedad difusa tienen más EPI (53 % versus 34 %) que aquellos con formas limitadas6.
Autoanticuerpos: 85 % de los individuos con anticuerpos antitopoisomerasa 1 (anti SCL 70) desarrollan EPID; se han visto involucrados otros anticuerpos (anti: U1-RNP, U3-RNP, Th/To y PM/ SCl), pero sin lograr establecer una asociación clara6.
Reflujo gastroesofágico (RGE): su asociación con el empeoramiento de la enfermedad ha sido controversial; no obstante, la mayoría de los estudios han encontrado una correlación positiva, apoyándose también en los datos histopatológicos de pacientes con fibrosis y RGE, en quienes se ha encontrado una distribución broncocéntrica con contenido intraluminal que se asemeja al fluido gástrico; con estos datos, se ha planteado un tratamiento intensivo del reflujo para evitar la progresión28.
Genes: en un ensayo que comparó la huella molecular en pulmón y fibroblastos pulmonares en 33 pacientes que llegaron al trasplante pulmonar, se encontraron patrones moleculares muy distintos en los fibroblastos de pacientes con ES y FPI: ambas entidades comparten solo 19 genes y se presentan diferencias en 59 y 77 patrones moleculares, respectivamente29. CXCL4 es uno de los genes más diferenciados y expresados en los estudios genómicos de ES y codifica para una proteína relacionada con los megacariocitos con un potente efecto antiangiogénico; su hallazgo puede incluso predecir la progresión de la enfermedad pulmonar30. Los polimorfismos de algunos locus en genes que codifican para el factor de crecimiento de los hepatocitos, el factor de crecimiento del tejido conectivo y CCL 18, entre otros, también se han asociado con el desarrollo de la enfermedad intersticial. Los anteriores hallazgos podrían enfocarse en lograr, en un futuro, una predicción más temprana de la enfermedad, así como en crear terapias dirigidas a un blanco específico.
¿CUÁNDO Y A QUIÉN TRATAR?
Según la recomendación de expertos, no todos los casos de EPID asociados a ES ameritan tratamiento; este se reserva para los pacientes en riesgo de rápido deterioro de la función pulmonar. En 2008 Goh y colaboradores23 publicaron un diagrama de flujo que tenía en cuenta la mortalidad, las pruebas de función pulmonar y la TCAR de 215 pacientes en tratamiento con esteroides, ciclofosfamida, azatioprina o micofenolato mofetil. De acuerdo con estos datos, se considera que un compromiso pulmonar mayor de 20 % según la TCAR se asocia de forma significativa con disminución rápida de la función pulmonar y muerte; esto se ha confirmado en los datos de otros estudios como el Scleroderma Lung Study 1 (SLS-1) en el cual los pacientes que más respondieron al tratamiento fueron los que tenían un compromiso mayor del 20 %31. En los pacientes en que es difícil establecer el porcentaje de compromiso pulmonar, una CVF menor de 70 % equivale a un compromiso por TCAR de más del 20 %23; estos dos parámetros son los indicados para iniciar el tratamiento.
¿CÓMO TRATAR?
Se han descrito medidas de soporte que se utilizan en casos individuales, entre ellas se encuentran:
Uso de oxígeno suplementario en pacientes con PO2 menor de 60 mm Hg.
Rehabilitación pulmonar.
Tratamiento del reflujo gastroesofágico.
Vacunación para influenza y pneumococo.
Profilaxis para Pneumocystis jirovecii en caso de tener un recuento de CD4 menor de 200/µL2.
Con respecto a la terapia farmacológica y modificadora de la enfermedad hay varias alternativas, entre ellas las siguientes (Tabla 3):
Inmunosupresores: ciclofosfamida, micofenolato mofetil, azatioprina.
Trasplante de médula ósea.
Trasplante pulmonar.
Tabla 3 Tratamiento de la enfermedad pulmonar intersticial en pacientes con esclerosis sistémica

IV: intravenosa
Ciclofosfamida: es la terapia más evaluada en ensayos con asignación aleatoria y que ha demostrado eficacia; el estudio más importante es el SLS publicado en 2006, que fue multicéntrico, doble ciego, controlado con placebo; incluyó 158 pacientes y el desenlace primario fue la mejoría de la CVF administrando ciclofosfamida oral a la dosis de 2 mg/kg/día por un año. El promedio de la diferencia absoluta a los 12 meses fue 2,53 %, con un intervalo de confianza del 95 % que atravesaba la unidad (0,28-4,79); a pesar de esto, cuando se compararon los pacientes que mejoraron con ciclofosfamida con los que recibieron placebo, se encontró una diferencia estadística y clínicamente significativa (49,3 % versus 26,4 %). Un compromiso en la TCAR mayor de 20 % y una CVF menor de 70 % fueron más frecuentes en los pacientes que mejoraron con ciclofosfamida31. No obstante, este resultado es transitorio, como se demostró en la extensión de dicho estudio en la cual se encontró que si bien el beneficio se prolongaba hasta 18 meses, la CVF se igualaba a los 2 años32, lo que recalca la importancia de continuar la inmunosupresión una vez terminada la fase inicial con ciclofosfamida.
Otro estudio que evaluó este fármaco fue el Fibrosing Alveolitis in Scleroderma Trial (FAST) en el que se utilizaron ciclofosfamida intravenosa por seis meses a la dosis de 600 mg/m2 /mes y prednisolona 20 mg interdiarios, seguidas de un régimen de mantenimiento con azatioprina a la dosis de 2,5 mg/kg/día. Al año no se encontraron diferencias significativas en comparación con el placebo. No obstante, es de anotar que este trabajo tuvo una muestra menor que la del SLS-1 y las medias de la CVF y el compromiso por TCAR eran del 80 % y menor del 20 %, respectivamente33 lo que, como se mencionó previamente, se asocia a menor respuesta a la ciclofosfamida.
Micofenolato mofetil y sódico: con este medicamento hay múltiples estudios sin asignación aleatoria, en su mayoría retrospectivos o de casos y controles, con resultados contradictorios. Aunque en términos generales la evidencia está a favor de usarlo, en muchas ocasiones este medicamento se administró combinado con otros inmunosupresoras lo que impide establecer el beneficio aislado del micofenolato34)(35)(36)(37)(38)(39)(40)(41)(42.
El SLS II comparó el micofenolato 1,5 gramos dos veces al día por dos años con la ciclofosfamida oral 2 mg/kg/día por un año y luego continuación con placebo. Si bien solo se cuenta con resultados parciales, estos muestran que ambos medicamentos tienen igual efectividad, pero con una tasa de abandonos y efectos adversos mayor en los pacientes tratados con ciclofosfamida43)(44.
Azatioprina: son pocos los estudios para destacar. Nadashkevich y colaboradores45 hicieron un estudio con 30 pacientes seguidos por 18 meses, con dosis de azatioprina de 2,5 mg/kg/día por 12 meses y luego mantenimiento con 2 mg/kg/día, comparada con ciclofosfamida oral 2 mg/kg/día por 12 meses y mantenimiento a la dosis de 1 mg/kg/día. Encontraron que la azatioprina era inferior tanto para preservar la CVF como la DLCO dejándola entonces como una opción para mantenimiento más que para el tratamiento inmunosupresor inicial.
Al revisar las guías de manejo, se observa que los expertos en el tema difieren tanto en el esquema de inducción como en el de mantenimiento46; esto quizás se explique por qué, por ahora, no hay información sólida sobre cuál sería la mejor opción terapéutica. Está claro que la ciclofosfamida detiene la progresión de la enfermedad en algunos pacientes y que, por el reporte preliminar del SLS II, el micofenolato mofetil podría ser una opción, siempre y cuando en cualquiera de los casos luego de inducir la remisión se continúe con terapia de mantenimiento para evitar perder el beneficio inicial obtenido.
Nuevos tratamientos
Se vienen utilizando otros tratamientos fuera de los convencionales con resultados promisorios en su mayoría, pero con las dificultades de tener poco tiempo de seguimiento y tamaño pequeño de la muestra. Entre ellos está el rituximab que ha demostrado eficacia en varios estudios; cabe resaltar, aunque solo fueron nueve pacientes, un trabajo de la cohorte EUSTAR con una buena respuesta en los individuos con ES y específicamente en pulmón32)(47.
El trasplante de médula ósea es otra de las opciones; están informados los resultados de dos estudios de pacientes con enfermedad pulmonar intersticial: el ASSIST fue el primero, con 19 pacientes seguidos por dos años; se comparó con ciclofosfamida 1 gramo/ m2 endovenoso mensual por seis meses y se encontró que tenían mejor CVF los trasplantados; sin embargo, a los dos años, esta venía en descenso, por lo que se ha puesto en duda el efecto a largo plazo48. En el ASTIS, 156 pacientes se asignaron a ciclofosfamida 750 mg/m2 cada mes por 12 meses o trasplante de médula ósea y se siguieron por 5,8 años; se encontró una mejoría mayor en los pacientes trasplantados, pero con una tasa alta de mortalidad por infecciones en el primer año49. En curso están el estudio STAT, sin brazo comparativo, de pacientes en quienes había fallado la terapia convencional50, y el SCOT que tiene brazo de trasplante y su control es ciclofosfamida a la dosis inicial de 500 mg/m2 en el primer mes y luego 750 mg/m2 mensual por 11 meses51.
Consecuente con la fisiopatología de la ES, se han utilizado agentes antifibróticos, bloqueando, por ejemplo, la expresión y señalización de TGF-β (pirfenidona, nintedanib), IL-6 (tocilizumab), anticuerpos anti-CTGF (factor de crecimiento del tejido conectivo), agonistas de PPAR-γ (receptor de peroxisoma proliferador activado, (rosiglitazona), estatinas, quinolonas e inhibidores de la trombina (dabigatrán), aún sin estudios clínicos controlados que demuestren su eficacia.
Finalmente, el trasplante pulmonar uni- o bilateral, que para muchos autores es prohibitivo, dada la concomitancia de la enfermedad pulmonar intersticial con reflujo gastroesofágico, lo que aumenta el riesgo de aspiración, bronquiolitis obliterante y con ello el rechazo del trasplante. No hay estudios controlados con asignación aleatoria, pero un metaanálisis reciente muestra que la supervivencia de estos pacientes puede ser igual a la de los trasplantados por otras causas siempre y cuando sean seleccionados cuidadosamente52.
CONCLUSIONES
La enfermedad pulmonar intersticial es en el momento la causa más importante de morbimortalidad en los pacientes con ES; es distinta de la fibrosis pulmonar, desde los puntos de vista molecular y clínico, así como en la respuesta a la terapia inmunosupresora. La decisión de tratamiento se fundamenta tanto en pruebas de función pulmonar como en las imágenes. La ciclofosfamida está soportada por la evidencia y requiere, luego de una fase de inducción de seis a 12 meses, continuar mantenimiento con otros inmunosupresores entre ellos el micofenolato que podría ser no inferior para la inducción. El resto de medicamentos mencionados podrían considerarse una alternativa ante la no respuesta. Finalmente, la mayoría de los pacientes, independientemente de la terapia farmacológica, requerirán otras medidas de soporte que mejoren su calidad de vida.

