











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkIntroducción
El rivaroxabán fue aprobado por la Administración de Alimentos y Medicamentos (FDA, por sus siglas en inglés) en 2008 y desde entonces se han realizado múltiples modificaciones que han permitido extender sus indicaciones autorizadas a diferentes patologías como el tromboembolismo pulmonar o venoso, el síndrome coronario agudo y la fibrilación 1. La molécula se cataloga de acuerdo con el Sistema de Clasificación Biofarmacéutica como de alta permeabilidad y baja solubilidad (clase II) 2.
El rivaroxabán es un anticoagulante oral altamente selectivo que actúa al inhibir directamente el factor Xa. Al bloquear la amplificación de la cascada de la coagulación e inhibir sus vías intrínseca y extrínseca evita la formación de trombina y, por consiguiente, de trombos. Dos mecanismos farmacocinéticos y farmacodinámicos lo hacen único en su clase: el primero es no implicar la vía de la antitrombina III y el segundo tener una vía de administración oral 3-5. Adicionalmente, no inhibe la trombina (factor II activado) y no se han demostrado efectos sobre las plaquetas 6.
El rivaroxabán se absorbe rápidamente y alcanza una concentración plasmática máxima en 2-4 horas, con una biodisponibilidad del 80-100 % para la dosis de 10 mg, y aproximadamente del 66 % en ayunas para la tableta de 15 mg 3-5. La ingesta de alimentos no afecta el área bajo la curva AUC o la concentración máxima (Cmáx) en la dosis de 10 mg 7. El rivaroxabán es un fármaco clase II (según la Agencia Europea de Medicamentos) con una biodisponibilidad del 66 % del comprimido de 20 mg, en ayunas 8. La AUC posprandial del comprimido de 20 mg aumenta en alrededor del 39 %, en comparación con los análisis en ayuno. Los comprimidos de 10, 15 y 20 mg han demostrado ser proporcionales en términos de AUC y Cmáx9. Tiene un aclaramiento de 10 L/H, es decir, bajo; su eliminación se produce con vidas medias (Vm) terminales de 5 a 9 horas en sujetos jóvenes y de 11 a 13 horas en sujetos de edad avanzada 9.
Dentro del perfil farmacocinético de un fármaco, la disolución se encuentra íntimamente relacionada con el proceso de absorción y es un determinante en la biodisponibilidad del medicamento y, por tanto, de la respuesta clínica. Los procesos de liberación in vitro permiten conocer los perfiles de disolución, las curvas de liberación contra el tiempo y, en algunos casos, las correlaciones in vitro-in vivo, con los que se puede determinar el comportamiento in vivo mediante un modelo in vitro10. Adicionalmente, los ensayos de disolución dan información sobre la biodisponibilidad de un fármaco en las diferentes dosis fabricadas y, en el caso de tener curvas similares en productos innovadores, demuestran el perfil de eficacia y seguridad.
La seguridad del rivaroxabán ha sido estudiada en estudios fase III en pacientes sometidos a cirugía ortopédica, cardiaca e infarto agudo de miocardio 11-14. En estos estudios, al menos el 67 % de los pacientes presentó reacciones adversas, de las cuales el 22 % se relaciona con el tratamiento. En pacientes tratados con 10 mg los eventos adversos son principalmente hemorrágicos (6, 8-12,6 %) y anemia (5,9 %). En pacientes tratados con 15 mg, 2/día o 20 mg/día, el 27,8 % tuvo eventos hemorrágicos y el 2,2 % anemia. Otros eventos adversos reportados (> 1/100 o 1/10) fueron el aumento de transaminasas, dolor en las extremidades, cefalea, hipotensión y prurito, entre otros 11-14.
En el presente estudio se analizó la equivalencia terapéutica de rivaroxabán (tabletas de 10 y 15 mg vs tabletas de 20 mg), con perfiles de disolución y un estudio in vivo comparativo de farmacocinética (entre rivaroxabán 20 mg en ayuno o posprandial).
Materiales y métodos
Medicamento
Se utilizó rivaroxabán (5-chloro-N-{[(5S)-2-oxo- 3-[4-(3-oxomorpholin-4-yl)phenyl]-1,3-oxazoli- din -5- yl]methyl}thiophene-2-carboxamide) 20 mg en comprimidos revestidos fabricados por Sanofi S. A. (lote: P0220318 y fecha de fabricación: 09/05/18) como producto de referencia, rivaroxabán 10 mg, comprimidos revestidos fabricados por Sanofi S. A. (lote: P0150318 y fecha de fabricación: 07/05/18) y rivaroxabán, comprimidos de 15 mg (lote: P0180318 y fecha de fabricación: 08/05/18) como medicamentos de estudio, para el estudio in vitro (perfiles de disolución: ver anexo 1).
En el análisis de bioequivalencia, se usó rivaroxabán comprimido revestido de 20 mg (lote 17100214) fabricado por Medley Farmacéutica Ltda. (medicamento de estudio), que se comparó con Xarelto®, rivaroxabán 20 mg comprimido revestido (Lote BXHL9A3) fabricado por Bayer Pharma A/G (medicamento de referencia).
Disolución in vitro
Se realizó un perfil comparativo de disolución para determinar la similitud entre tres compuestos de rivaroxabán (10 y 15 mg vs 20 mg). Se utilizó cromatografía líquida [columna C8, 250x4,6 mm, 5 μm; λ: 249 nm; flujo: 1.0 ml/min; temperatura: 25 °C; volumen de inyección: 50 μL; fase móvil: agua: acetonitrilo (55:45 v/v)]. Se realizaron evaluaciones de 12 unidades de dosificación por cada medio y muestreos en diferentes intervalos de tiempo (5, 10, 15, 30, 45 y 60 minutos). El estudio del perfil de disolución se realizó de acuerdo con un método previamente validado (anexo 1) y se ejecutó en tres soluciones de rivaroxabán en agua-acetonitrilo (20:80 v/v) a una concentración de 0,1 mg/ml; de esta se tomaron réplicas de los medios de disolución (pH 1,2, 4,5 y 6,8) a diferentes niveles de concentración (1 %, 5 %, 25 %, 50 %, 75 %, 100 % y 120 %). Las muestras fueron filtradas por un filtro de membrana de celulosa regenerada con tamaño de poro de 0,45 ^m y un diámetro de 13 mm e inyectadas directamente al cromatógrafo.
Las curvas de calibración se obtuvieron de la ecuación recta para determinar el porcentaje disuelto de principio activo en cada medio de disolución y en cada tiempo de muestreo. Para la comparación de los datos en los perfiles de disolución se usó un factor de similitud (f2). Se determinó similitud con valores de 50 y cercanos a 100. Se usaron valores de disolución media para estimar el factor de similitud f2 y para calcular los datos medios. El coeficiente de variación en los puntos de tiempo no debía ser mayor del 20 % a los 10 minutos y del 10 % en otros tiempos. En caso de que ambos productos se disolvieran en el 85 % o más de la cantidad declarada de principio activo en 15 minutos o menos, la comparación de los perfiles no sería necesaria 15,16. La cuantificación de rivaroxabán se basó en la construcción de curvas de calibración en cada medio de disolución, que se sometieron a un análisis de regresión lineal.
Estudio de bioequivalencia
Se realizó un ensayo clínico abierto, aleatorizado, cruzado, con dos tratamientos, en dos periodos (ayuno y posprandial) y dos secuencias. Se incluyeron sujetos entre los 18 y 50 años, sin antecedentes médicos de importancia, corroborados mediante la historia clínica y confirmados por pruebas de laboratorio [ECG, Hemograma, Tiempo de protrombina (TP), tiempo de tromboplastina parcial activada (TTPa), aclaramiento de creatinina, ácido úrico, urea, colesterol total y triglicéridos, (AST/TGO), (ALT/TGP), fosfatasa alcalina, bilirrubina total y fracciones, proteínas totales, albúmina, creatinina, glucosa en ayunas, parcial de orina, serología, análisis serológico para: hepatitis B, hepatitis C y virus de la inmunodeficiencia humana y β-HCG para mujeres], con índice de masa corporal entre 18,5 y 29,9 kg/m2 y con tiempos de coagulación y aclaramiento de creatinina dentro de límites normales, que comprendieran y aceptaran el ingreso al estudio por medio de un consentimiento informado. Los sujetos fueron analizados con un intervalo mínimo de confinamiento de siete días. Se administró un comprimido de rivaroxabán revestido de 20 mg fabricado por Medley Farmacéutica Ltda. (medicamento de estudio), que se comparó con un comprimido de Xarelto®, rivaroxabán 20 mg, revestido, fabricado por Bayer Pharma A/G, administrado por vía oral, en dosis única (medicamento de referencia).
Los voluntarios fueron confinados en un único grupo (de 1-24 y 25-48 para el grupo en ayuno y de 1-18 y 19-36 para el grupo posprandial) entre las 19:00 y las 21:26 para el grupo en ayunas y las 19:11 y las 20:59 para el grupo posprandial del día anterior al inicio de administración del fármaco, y con un tiempo de confinamiento de 12 horas preadministración y hasta 24 horas posadministración.
Evaluación en ayuno
Los sujetos permanecieron en ayuno absoluto desde las 22 horas del inicio del confinamiento. Luego de 2 horas de la administración del medicamento se les ofrecieron 200 ml de agua y a partir de este horario, se permitió ingesta de agua a libre demanda. No se ofrecieron bebidas con xantinas. Se sirvió un refrigerio a las 21 horas, almuerzo a las cuatro horas posmedicamento, merienda a las ocho horas, cena a las diez horas y refrigerio a las 14 horas posmedicamento. Se recolectaron 22 muestras (ver anexo 2) para la cuantificación plasmática, en tubos con anticoagulante EDTA, centrifugados a 3000 rpm por 10 minutos, almacenados a -70 °C.
Evaluación posprandial
Los sujetos permanecieron en ayuno absoluto desde las 21:30 horas del confinamiento. Treinta minutos antes de la administración de la medicación se ofreció dieta hipercalórica. La ingesta de agua y alimentos siguió el mismo esquema de alimentación que en los pacientes del brazo A. Se recolectaron 21 muestras (ver anexo 3) con el mismo esquema del brazo A.
Seguridad y tolerancia
La seguridad y tolerancia al medicamento se determinó en todos los momentos del estudio usando el reporte de síntomas, exámenes paraclínicos y seguimientos clínicos.
Análisis de los datos
Para la cuantificación de rivaroxabán se usó cromatografía líquida acoplada a la espectrometría de masas en modo MS/MS con patrón interno de rivaroxabán-d4. Se determinó linealidad por el método analítico en el rango de concentración de 1,0-6000,0 ng/ml. La cuantificación se realizó por medio del uso de razón de áreas de los picos de rivaroxabán (MRM436,24>144,85)/rivaroxabán-d4 (MRM 440,38/144,98).
Resultados
Disolución in vitro
Se evidenció que los perfiles de disolución de rivaroxabán de 10 y 15 mg, en los medios de disolución evaluados (pH 1,2, 4,5 y 6,8) fueron comparables con rivaroxabán 20 mg (con un perfil de disolución > 50 en todos los medios). Las figuras 1 a 3 muestran los perfiles de disolución de rivaroxabán en diferentes medios buffers, en los seis tiempos de seguimiento, como se muestra a continuación:
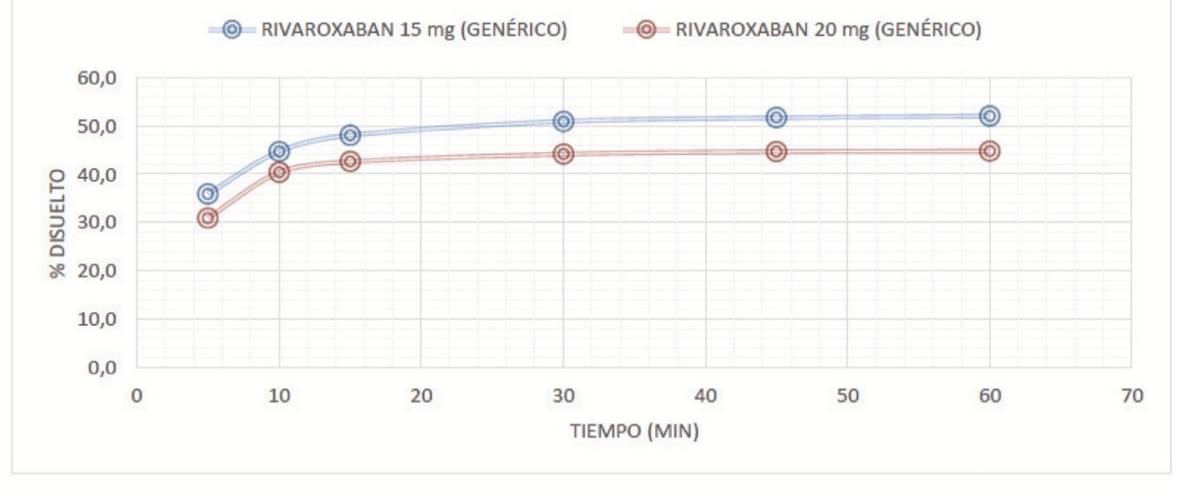
Fuente: elaboración propia.
Figura 1 Perfil de disolución comparativo de rivaroxabán 15 mg vs rivaroxabán 20 mg en buffer pH 1,2

Fuente: elaboración propia.
Figura 2 Perfil de disolución comparativo de rivaroxabán 15 mg vs rivaroxabán 20 mg en buffer pH 4,5

Fuente: elaboración propia.
Figura 3 Perfil de disolución comparativo de rivaroxabán 15 mg vs rivaroxabán 20 mg en buffer pH 6,8
Estudio de bioequivalencia
Se incluyeron 48 voluntarios en el grupo en ayunas y 28 en el grupo posprandial. Las características basales entre los grupos fueron similares (tabla 1). No hubo casos de desistencia/exclusión o no inclusión de los voluntarios en el estudio en ayunas. Sin embargo, en el grupo posprandial un paciente desistió de continuar en el estudio antes de la administración del medicamento (motivos personales) y otro fue excluido del estudio durante el periodo de wash out. Se reportaron desviaciones del protocolo por recolección de muestras en un intervalo de +2 horas en 47 periodos, así como desviaciones del protocolo en el consumo de alimentos en 183 casos para los pacientes del grupo de ayuno y 10 casos de desviaciones del protocolo en los pacientes del grupo posprandial en el intervalo de recolección de muestra de +2 horas. De igual forma nueve sujetos en el periodo 1 y 10 en el período 2 tuvieron una recolección de 24 horas y alta al confinamiento antes de lo previsto por motivos personales. Adicionalmente, 52 periodos presentaron alteraciones del protocolo en la administración de alimentos en el grupo posprandial.
Tabla 1 Características de los pacientes en el estudio
| Variable | Ayuno (n=48) | Posprandial (n=24) |
|---|---|---|
| Edad | 18-46 | 18-46 |
| Índice de masa corporal | 19,5-29,7 kg/m2 | 19,4-29 kg/m2 |
| Género (femenino) | 50 % | 53 % |
Fuente: elaboración propia.
El rivaroxabán de referencia y el de estudio, tanto en pacientes en ayuno como en pacientes en periodo posprandial, mostró patrones de bioequivalencia comparables, al tener valores extremos del intervalo de confianza del 90 % de la razón de los promedios geométricos (AUC0-t prueba/AUC0-t referencia y Cmáx prueba/Cmáx referencia), mayores al 80 % y menores al 125 %.
Tabla 2 Valores de la Cmáx, auc0-1, auc0-inf para rivaroxabán
| Ayuno | Posprandial | |||||
|---|---|---|---|---|---|---|
| Razón (%) | Poder (%) | Valor p* (%) | Razón (%) | Poder (%) | Valor p* (%) | |
| Cmáx | 100,77 | 99,99 | 83,53 | 110,63 | 99,84 | 70,41 |
| (IC95 % 94,24-107,76) | (IC95 % 102,39-119,54) | |||||
| AUC0-t | 100,65 | 100,00 | 92,52 | 104,18 | 100,00 | 57,85 |
| (IC95 % 96,13-105,39) | (IC95 % 98,70-109,97) | |||||
| AUC0-inf | 98,92 (IC95 % 93,77-104,35) | 100,00 | 59,13 | 103,65 (IC95 % 98,44-109,12) | 100,00 | 57,67 |
Fuente: elaboración propia.
Adicionalmente, en los pacientes en ayuno la evaluación/comparación de la variabilidad de las formulaciones mediante los intervalos de confianza del 90 % de las razones σWT/ σWR para cada uno de los parámetros Cmáx, AUC0-t y AUC0-inf mostró que los límites superiores de los IC90 % fueron < 2,5 (tabla 3).
Eventos adversos
El 61,9 % de los eventos adversos se advirtió en los pacientes con el medicamento de referencia, mientras que el 38,1 % en el medicamento de estudio. No se presentaron eventos adversos fatales, serios, de especial interés o que tuvieran una relación con el medicamento y conllevaran al abandono del fármaco de estudio o del estudio (tabla 4).
Tabla 4 Eventos adversos durante el estudio
| Periodo | E/R* | Evento adverso | Evento grave | Esperado/No esperado | Intensidad | Causalidad | Evolución |
|---|---|---|---|---|---|---|---|
| 1 | E | Cefalea | No | Esperado | Leve | Posible | Recuperado |
| 2 | R | Cefalea | No | Esperado | Leve | Posible | Recuperado |
| 3 | R | Alteración laboratorio (urinario) | No | Esperado | Leve | Posible | Recuperado |
| 1 | E | Malestar pospunción | No | No esperado | Leve | No relacionado | Recuperado |
| 2 | R | Malestar pospunción | No | No esperado | Leve | No relacionado | Desconocido |
| 3 | R | Alteración laboratorio (urinario) | No | Esperado | Leve | Posible | Desconocido |
| 4 | R | Alteración laboratorio (hemograma) | No | Esperado | Leve | Posible | Desconocido |
| 5 | R | Alteración laboratorio (urinario) | No | No esperado | Leve | Improbable | Desconocido |
| 1 | E | Alteración laboratorio (urinario) | No | Esperado | Leve | Definido | Recuperado |
| 2 | R | Cefalea | No | Esperado | Leve | Posible | Recuperado |
| 1 | E | Cefalea | No | Esperado | Leve | Posible | Recuperado |
| 2 | R | Cefalea | No | Esperado | Leve | Posible | Recuperado |
| 2 | R | Alteración laboratorio (leucograma) | No | No esperado | Leve | Improbable | Desconocido |
| 1 | R | Malestar pospunción | No | No esperado | Leve | No relacionado | Recuperado |
| 2 | E | Cefalea | No | Esperado | Leve | Posible | Recuperado |
| 2 | E | Alteración laboratorio (urinario) | No | Esperado | Leve | Posible | Desconocido |
| 2 | R | Alteración laboratorio (transaminasas) | No | Esperado | Leve | Posible | Desconocido |
| 2 | R | Alteración laboratorio (urinario) | No | Esperado | Leve | Posible | Desconocido |
| 1 | E | Cefalea | No | Esperado | Leve | Posible | Recuperado |
| 2 | R | Cefalea | No | Esperado | Leve | Posible | Recuperado |
| 2 | R | Cefalea | No | Esperado | Leve | Posible | Recuperado |
| 2 | R | Alteración laboratorio (colesterol total) | No | No esperado | Leve | Improbable | Desconocido |
| 2 | R | Alteración laboratorio (leucograma) | No | No esperado | Leve | Improbable | Desconocido |
| 1 | E | Cefalea | No | Esperado | Leve | Posible | Recuperado |
| 2 | R | Alteración laboratorio (leucograma) | No | No esperado | Leve | Improbable | Desconocido |
| 2 | R | Alteración laboratorio (urinario) | No | Esperado | Leve | Posible | Desconocido |
| 2 | E | Alteración laboratorio (urea) | No | Esperado | Leve | Posible | Recuperado |
| 2 | E | Alteración laboratorio (eritrograma) | No | Esperado | Leve | Posible | No recuperado |
| 1 | E | Cefalea | No | Esperado | Leve | Posible | Recuperado |
| 2 | R | Alteración laboratorio (leucograma) | No | No esperado | Leve | Improbable | Recuperado |
| 2 | E | Alteración laboratorio (hemograma) | No | Esperado | Leve | Posible | Desconocido |
| 1 | E | Cefalea | No | Esperado | Leve | Posible | Recuperado |
| 2 | E | Cefalea | No | Esperado | Leve | Posible | Recuperado |
| 2 | E | Cefalea | No | Esperado | Leve | Posible | Recuperado |
| 2 | E | Alteración laboratorio (leucograma) | No | No esperado | Leve | Improbable | Recuperado |
| 2 | E | Alteración laboratorio (triglicéridos) | No | No esperado | Leve | Improbable | Desconocido |
| 2 | R | Alteración laboratorio (hemograma) | No | No esperado | Leve | Improbable | Desconocido |
| 2 | R | Malestar pospunción | No | No esperado | Leve | No relacionado | Recuperado |
| 1 | R | Malestar pospunción | No | No esperado | Leve | No relacionado | Recuperado |
| 1 | R | Malestar pospunción | No | No esperado | Leve | No relacionado | Recuperado |
| 1 | R | Caída de moto | No | No esperado | Leve | No relacionado | Recuperado |
| 1 | R | Caída de moto | No | No esperado | Leve | No relacionado | No recuperado |
| 1 | R | Malestar pospunción | No | No esperado | Leve | No relacionado | Recuperado |
*E = medicamento de estudio, R = medicamento de referencia.
Fuente: elaboración propia.
Discusión
El presente estudio evidenció parámetros de disolución para rivaroxabán de 10 y 15 mg, en diferentes medios estudiados, comparables con rivaroxabán 20 mg (producto de referencia). Se demuestra que el rivaroxabán de 10, 15 y 20 mg tiene un perfil de disolución similar al medicamento de referencia en el rango de pH fisiológico. Lo anterior determina una absorción equivalente de las diferentes dosis de estudio en comparación con el patrón de referencia, que se traduce en un efecto terapéutico equivalente, según lo determina el supuesto fundamental de la bioequivalencia.
Al comparar las formulaciones de estudio con la de referencia, se evidenció que la media de la Cmáx, el AUC0-1 y el AUC0-inf para el rivaroxabán de estudio son equivalentes al medicamento de referencia, cumpliendo los criterios de bioequivalencia al tener una Cmáx entre un ± 30 % (70-130 %) y AUC entre un ± 20 % (80-120 %) 17-19. Según estos resultados, se puede asumir que, tratándose de medicamentos bioequivalentes, tendrán el mismo efecto farmacológico y, por tanto, son terapéuticamente equivalentes y completamente intercambiables 18.
Además, el reporte de eventos adversos en nuestro estudio mostró que el síntoma más frecuente posiblemente relacionado con el medicamento de prueba fue cefalea (n = 13), sin que se presentaran eventos fatales o que llevaran a la suspensión del medicamento. La frecuencia de estos eventos en el medicamento de referencia fue menor en el medicamento de estudio y no se presentó sangrado, reportado como el evento adverso más frecuente (> 5 %) en el tratamiento con rivaroxabán 1. Lo anterior demuestra un adecuado perfil de seguridad, sin la presencia de eventos adversos serios o fatales.
El uso de estudios bioanalíticos de biodisponibilidad previamente validados en la literatura médica 20, así como un análisis realizado acorde con la normativa internacional para estudios de disolución y de bioequivalencia (intervalo de confianza del 90 %). Para la biodisponibilidad relativa debe estar dentro de un rango de bioequivalencia de 80,00 a 125,00 % y valor F2 mayor a 50 para perfiles de disolución 17-19 que garantizan la confiabilidad de los datos obtenidos en el presente estudio. Es importante resaltar que la bioequivalencia en medicamentos de bajo costo al ser comparados con la molécula original, permite ampliar indirectamente el acceso del medicamento a la población, en razón a los precios competitivos.
Adicionalmente, los hallazgos reportados se soportan en las normas locales e internacionales, según las cuales, a partir de la demostración de equivalencia farmacéutica y los resultados de los estudios de biodisponibilidad, se puede confirmar si existe o no un perfil de eficacia y seguridad del medicamento de estudio similar al medicamento de referencia 21,22. De igual manera, los resultados descritos pueden ser sustitutivos para los criterios de análisis clínico de eficacia y seguridad del estudio en el marco de un ensayo clínico 18.
Conclusiones
Según las normativas internacionales de bioequivalencia, cuando existe un estudio in vivo, con la mayor concentración de un rango de concentraciones, las concentraciones menores se evalúan por medio de la bioexención por proporcionalidad de dosis (estudio in vitro)
Como evidencia el presente estudio, en el rango de pH fisiológico, la formulación de rivaroxabán de 10 y 15 mg en tabletas recubiertas fabricadas por Sanofi presenta una cinética de disolución similar a la formulación de rivaroxabán 20 mg tabletas recubiertas fabricadas por Sanofi, establecida a través del cálculo de un valor F2 mayor a 50 para los perfiles de disolución. Con el estudio de bioequivalencia/biodisponibilidad se determina que existe bioequivalencia entre la formulación de referencia [rivaroxabán 20 mg (Xarelto®)] y la de estudio [rivaroxabán 20 mg] fabricado por Medley Farmacéutica Ltda., tanto en condición de ayuno como posprandial, infiriendo así que los medicamentos tendrían el mismo efecto farmacológico y son equivalentes desde el punto de vista terapéutico.

