











 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
Permalink
INTRODUCTION
Tachykinins are a diverse family of pleiotropic neuropeptides which exhibit significant neuromodulating properties. Several tachykinins have been identified in humans, some of which include substance P, hemokinin-1, endokinin-A, endokinin-B, neurokinin-A, and neurokinin-B [1]. These neuroactive compounds act through an array of receptors encoded in the human genome and which are actively involved in numerous neuroendocrine circuits [2, 3].
Tachykinin/neurokinin 3 receptor (NK3R) exhibits a high affinity for neurokinin-B [1] and when activated, it displays cholinergic modulation and endocrine reproductive regulation mediated by neurohormonal action on the hypothalamus [4]. In the arcuate/infundibular nucleus (ARC), neurokinin-B is co-expressed with kisspeptin and dynorphin by KNDy neurons which project to GnRH-releasing cells and thermoregulatory centers. During menopause, KNDy neurons hypertrophy in response to the absent feedback of estrogen, therefore there is an augmented firing rate towards NK3 R-expressing hypothalamic neurons either within the ARC nucleus (GnRH neurons) or towards the preoptic area (thermoregulatory center and GnRH neurons) [5].
Soon after menopause onset, induced-hypertrophy stimulates KNDy neurons to release kisspeptin leading to GnRH increased secretion and further FSH and LH serum elevation which is commonly seen in the initial phases of menopause. Simultaneously, greater stimulation of the preoptic area via neurokinin-B signaling results in autonomic activation causing the frequent menopause-associated vasomotor symptoms commonly known as hot flashes [5] (figure 1).

Figure 1 Neuroendocrine circuits involved in hot flashes onset. Postmenopausal hormonal decrease results in the blockage of E2 hypothalamic downregulation of ARC neurons, therefore, inducing KNDy hypertrophy with increased excitability. Augmented release of KissP stimulates GnRH secretion towards the median eminence and results in FSH and LH raised levels. POA neurons are stimulated via NKB-NK3R leading to vasodilation and sweating mediated by autonomic outputs. E2: Estradiol; ARC: arcuate nucleus; POA: Preoptic area; KNDy: Kisspeptin-NeurokininB-Dy-norphin neuron; NKB: Neurokinin-B; NK3R: Neurokinin 3 receptor; KissP: Kisspeptin; KisslR/ GPR54: Kisspeptin receptor. Source: Own elaboration.
As a result, NK3Rhas become a suitable target for drug development in disorders such as schizophrenia, and non-hormonal therapy during menopause [6, 7]. Around 2005, the first non-peptidic NK3R antagonists were reported, Osanetant (SR142801) and Talnetant (SB223412), both developed as antipsychotics [8]. More recendy, a reduction of hot flashes was identified when inhibiting NK3R, giving rise to compounds for vasomotor symptoms therapy which are currently under study in humans and animal models [9]. NK3R antagonists are shown in figure 2.

Figure 2 Molecular structure of NK3R antagonists. Compounds advised for schizophrenia (Talnetant, Osanetant) and hot flashes therapy (Pavinetant, Fezolinetant, SB-222.200, SB-218.795). Source: Structures taken from [10].
Osanetant and Talnetant were tested for schizophrenia treatment and showed promising results in hospitalized patients, however, despite their efficacy, both drugs' clinical trials were finally suspended due to their poor pharmacokinetic profile [8]. On the other hand, several NK3R antagonists, yet not intended for schizophrenia but hot flashes therapy, have recently been raised as potential therapeutic agents. Pavinetant and Fezolinetant are both currently under clinal trials and have shown a significant reduction of hot flashes [11], with Fezolinetant presently undergoing Phase IIb [12]. Reduction of hot flashes has also been reported for compounds tested in animals, among them, SB-222.200 and SB-218.795 show high affinity for hypothalamic NK3R and exhibited promising results in preclinical trials [9].
NK3R antagonists have shown encouraging outcomes when studying their efficacy whether it is for schizophrenia or hot flashes reduction. Regardless of their distinct therapeutic effect, these compounds are orally-delivered drug candidates that target structures within the nervous system, therefore, assessing their pharmacokinetics is of great importance when evaluating their potential as drugs. Determining the pharmacokinetic properties of novel compounds requires the design of wasteful in vivo assays, as a result, in silico experimentation emerges as a promising strategy to accelerate drug filtering by providing several compound screening methods ranging from structure-activity analysis to toxicology and pharmacokinetic studies [13]. Physicochemical and ADME/T parameters (absorption, distribution, metabolism, excretion, and toxicity) can be predicted based on topological and molecular descriptors determined in bioinformatic platforms such as ADME/Tlab and SwissADME [10, 14].
As previously mentioned, Osanetant and Talnetant were discontinued as a result of their poor pharmacokinetics, consequently, novel NK3R antagonists are expected to exhibit superior drug-likeness in order to achieve better outcomes in future trials. This study aims to develop a first-time reported pharmacokinetic profiling of NK3R antagonists. By comparing the pharmacokinetic profile of Osanetant & Talnetant and novel NK3R antagonists, we intend to identify enhanced drug-like properties for Fezolinetant, Pavinetant, SB-222.200, and SB-218.795. Results derived from this investigation could help to elucidate optimal physicochemical and pharmacokinetic properties in current drug candidates for hot flashes reduction as well as to point out important features in possible scaffolds for future lead-compounds synthesis of potential drugs with NK3R antagonistic activity.
METHODOLOGY
NK3R antagonists, selection and structure retrieval
Compounds with hot flashes reducing activity were retrieved from Muñoz et al. [9]. NK3R antagonists under study were divided into three groups; schizophrenia drugs for which clinical trials were suspended [8], drugs under current testing in humans which have shown to ameliorate menopause vasomotor symptoms [12, 15], and animal-tested NK3R antagonists with a reported lessening of hot flashes [16, 17]. Carried out in silico analysis required the SMILES chemical identifiers which were taken from DrugBank (https://www.drugbank.com) and PubChem (https://pubchem.ncbi.nlm.nih.gov).
Physicochemical properties prediction
The assessment of physicochemical properties was based on predicted values calculated in the SwissADME platform (http://www.swissadme.ch). Analyzed parameters were those stated as crucial for suitable drug-like compounds [18]; lipophilicity (Log P), solubility (Log S), size (molecular weight), polarity (topological polar surface area), unsaturation (fraction of sp3 carbons) and flexibility (number of rotatable bonds). Retrieved values were plotted on a bioavailability radar chart [10].
Drug-likeness rules compliance
Drug-like properties for all NK3R antagonists were evaluated through their compliance with four drug-likeness rules: Lipinski, Ghose, Veber, and Varma [19-21]. Results were obtained from ADMETlab and further on analyzed as percentage values according to the number of violations for each assessed rule.
In silico ADME/T evaluation
The pharmacokinetic profile of each NK3R antagonist was determined through the calculation of absorption, distribution, metabolism, excretion and toxicity (ADMET) performed in the ADMETlab platform (http://ADMET.scbdd.com) [22]. Values retrieved from ADMETlab were based on Smiles inputs and were predicted with a 67% to 93% accuracy, depending on the estimated parameter.
Absorption & Distribution
Human gastrointestinal absorption (HIA), membrane permeability (Caco-2 permeability) and P-glycoprotein substrate likeliness were determined to evaluate absorption properties. In addition, distribution assessment was carried out by blood-brain barrier (BBB) diffusion and bioavailability prediction. Distribution results were reported as probability values for BBB diffusion and the achievement of a systemic circulating fraction of 20% and 30% when orally consumed.
Metabolism & Excretion
Interactions with cytochrome P450 enzymes, both as substrates and inhibitors, were predicted for the NK3R antagonists. Studied CYP450 included: CYP1A2, CYP2C19, CYP2C9, CYP2D6 and CYP3A4. Results were reported as probability values for inhibition and substrate interactions.
Toxicity
Toxicity evaluation was based on three estimated parameters: long-term human hepatotoxicity (H-HT), drug-induced liver injury (DILI) and probability of hERG blockage.
Data analysis
The aforementioned groups were compared in terms of physicochemical properties, drug-likeness compliance, and ADME/T predicted values. Statistical analysis was carried out by a one-way Anova test and taking into consideration significant differences with a probability of less than 0.05 (α=0.05).
RESULTS AND DISCUSSION
Physicochemical evaluation
Predicted values for lipophilicity, solubility, polarity, flexibility, size and unsaturation are presented in table 1. The overall results from physicochemical assessment did not report significantly high or low values for most of the studied compounds, still, some exhibited a greater number of non-suitable features. Statistical analysis revealed no significant difference (p>0.05) between the three groups in terms of physicochemical properties.
Table 1 Physicochemical properties of NK3R antagonists. Lipophilicity (Log P), Solubility (Log S), Polarity (TPSA), Flexibility (N.° RB), Size (MW) and Unsaturation (Fraction C sp3).
| Property | Suspended trial | Human tested | Animal tested | |||
|---|---|---|---|---|---|---|
| Talnetant | Osanetant | Pavinetant | Fezolinetant | SB-222.200 | SB-218.795 | |
| Log P (XLOGP3) | 5.75* | 6.97* | 5.07* | 1.56 | 5.92* | 4.75 |
| Log S (ESOL) | -6.00 | -7.65* | -5.85 | -3.32 | -6.09* | -5.37 |
| TPSA (Á2) | 62.22 | 43.86 | 96.54 | 105.04 | 41.99 | 68.29 |
| N.° RB | 6.00 | 10.00* | 8.00 | 3.00 | 6.00 | 7.00 |
| MW (g-mol-1) | 382.45 | 606.62* | 459.56 | 358.39 | 380.48 | 396.44 |
| Fraction C sp3 | 0.12* | 0.43 | 0.15* | 0.31 | 0.15* | 0.08* |
*Exceeding values. MW: Molecular weight; TPSA: Topological polar surface area; RB: Rotatable bonds.
Results from the physicochemical evaluation suggest that there are slight variations among the studied compounds which, in turn, affect their drug-like properties. In the first place, schizophrenia (suspended) drugs exhibited exceedingly high values of lipophilicity, insolubility, flexibility, and molecular weight. Human-tested drugs, on the other hand, showed only two violations concerning unsaturation and lipophilicity. Similar results were found in animal-tested compounds, which exhibited three violations for lipophilicity, solubility and unsaturation. For easier visual analysis, assessed properties for each compound are displayed as bioavailability radar charts (figure 3).

Figure 3 Bioavailability radar chart. Red-colored area: suitable drug-like properties values. Lipo: log P; Size: Molecular weight; Polar: Topological polar surface area; Insolu: log S; Insatu: Fraction of sp3 carbons; Flex: N.° of rotatable bonds. Plotted data within the red area represent suitable properties for drug-like compounds. Limit values are: -0.7 < logP < 5,150 g-mol1 < MW < 500 g-mol 20 Å2 < TPSA < 130 Å 2, -6 < log S <0, 0.25 < Fraction С sp3 < 1 and 0 < N.° RB < 9. Source: [10].
All six compounds under study exhibited adequate polarity (TPSA), yet several deviations were found in terms of the five sparing properties. Obtained results show that Talnetant has undesirable lipophilicity and unsaturation. Lipophilicity is a crucial parameter in drug-likeness evaluation; while low values indicate poor lipid bilayer permeability, exceeding values suggest, as in the case of Talnetant, that this compound would have poor blood transport and would tend to deposit in lipid-abundant tissues [23]. In terms of unsaturation, the fraction of sp3 carbons alludes to the spatial complexity and is tightly connected to solubility [24], hence Talnetant is predicted to exhibit properties that compromise its solvation, blood transport, and plasma distribution. Osanetant, on the other hand, was reported to be the least suitable drug-like compound, with violations for flexibility, size, lipophilicity, and solubility. Absorption and distribution are compromised by inappropriate values of solubility and lipophilicity [25], additionally, unsuitable size also conveys poor permeability and absorption, and indicates low synthetic accessibility [26]. Optimal flexibility, on the other hand, has recently shown to be crucial in drugs that target proteins (such as NK3R) given their continuous conformational changes [27], hence inadequate flexibility might compromise ligand-target affinity.
In regards to human-tested drugs, only Pavinetant reported slight violations for lipophilicity and unsaturation, whereas Fezolinetant showed no violations at all. Compounds with satisfactory values for all physicochemical parameters are predicted to exhibit superior pharmacological qualities [28]. On the other hand, animal-tested compounds displayed better properties than suspended drugs, yet not better than human-tested. Reported violations included lipophilicity, solubility, and unsaturation for SB-222.200, and unsaturation for SB-218.795.
When comparing novel NK3R antagonists undergoing clinical trials for hot flashes reduction and NK3R antagonists for which trials were suspended, results show that human-tested compounds exhibit enhanced drug-like properties. Fezolinetant showed more satisfactory results when compared to Talnetant and Osanetant, indicating that, in terms of physicochemical descriptors, Fezolinetant displays advantages that might play an important role when evaluating bioavailability and effectiveness following oral administration [29, 30].
Drug-likeness compliance
Based on predicted physicochemical properties, several guidelines have been developed in order to assess compounds in terms of drug-likeness. These are known as drug-likeness rules, in which different criteria are evaluated for each filter. In this study, we analyzed the compliance with Lipinski's rule of 5, Ghose filter, Veber's rules, and Varma's rules. An array of chemical descriptors are assessed by these guidelines, including molecular weight (MW), lipophilicity (Log P), hydrogen-bond acceptors and donors, molar refractivity (MR), number of atoms, number of rotatable bonds, polar surface area (TPSA), and solubility at pH=7.4 (Log D) [31, 32].
Results obtained for the three groups under study were reported as a mean percentage of compliance with each set of rules, hence a higher value represented a lower number of violations. The statistical analysis showed no significant difference (p>0.05) among the three groups in terms of drug-likeness, yet Talnetant and Osanetant show less compliance with Lipinski, Ghose, and Varma rules when compared to Fezolinetant, Pavinetant, SB-222.200, and SB-218.795. Mean compliance percentage values are shown in figure 4.
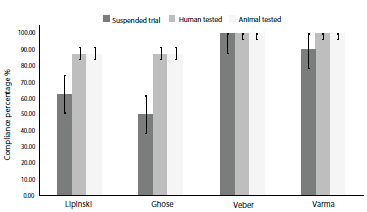
Figure 4 Drug-likeness rules mean compliance percentage for each of the four assessed guidelines ± SEM (standard error of the mean).
Although an in silico drug-likeness assessment is limited when classifying a molecule as a suitable drug [33], these guidelines provide valid information of proper pharmacokinetic behavior when administered orally [14, 34]. In concordance with our results, up to date carried out clinical trials showed slightly better outcomes for Fezolinetant when compared to Talnetant and Osanetant [9] and given the data obtained from this investigation, our findings suggest superior outcomes in more advanced clinical trials based on in silico pharmacokinetics prediction.
In silico ADME/T analysis
The assessment of the pharmacokinetic parameters for the NK3R antagonists was carried out in the ADMETlab platform. Initial analysis was based on BBB and GI absorption (figure 5), membrane permeability, and P-glycoprotein substrate properties (table 2).
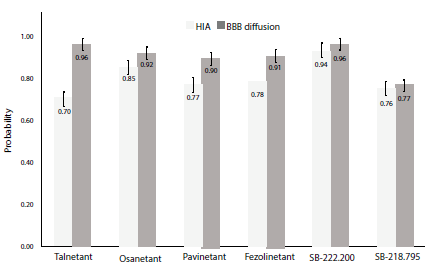
Figure 5 Human gastrointestinal absorption and blood-brain barrier diffusion probability ± SE. HIA: Human intestinal absorption; BBB: blood-brain barrier; SE: standard error.
Human intestinal absorption was predicted as stated in [35] with an accuracy of 78%. ADMETlab threshold for absorbable compounds states a cut-off point of 50%, meaning that compounds with probability values higher than 0.50 are classified as GI absorbable. All six compounds exhibited a human intestinal absorption value ≥70%, with SB-222.200 showing the highest probability. Smith et al. [36] determined that SB-222.200 was highly absorbable when orally administered to male rats, and complemented with this study, results suggest that SB-222.200 could exhibit high human GI absorption. Despite lower values for Fezolinetant absorption when compared to Talnetant and Osanetant, previous studies have shown that oral administration results in successful reduction of hot flashes [37], thus this degree of difference in GI absorption may not be significant enough to argue lower effectiveness of novel Fezolinetant.
Blood-brain barrier (BBB) diffusion evaluation show a 92% accuracy based on calculations proposed in [38]. Five compounds reported values ≥90%, except for SB-218.795. However, all six molecules were above the 0.50 threshold, suggesting suitable BBB diffusion. Compounds targeting structures in nervous tissue are required to have an equilibrium between solubility and lipophilicity balanced enough to cross the blood-brain barrier [39], our study shows that all six compounds exhibit appropriate BBB diffusion properties. Nevertheless, it should be noted that ionization states and stereochemical modifications are not considered when predicting this parameter. Further on, Caco-2 permeability [40] was evaluated parallel to P-glycoprotein substrate probability [41] (table 2).
Table 2 Caco-2 permeability and P-glycoprotein (Pgp) substrate probability of NK3R antagonists.
| Parameter | Suspended trial | Human tested | Animal tested | |||
|---|---|---|---|---|---|---|
| Talne-tant | Osane-tant | Pavine-tant | Fezoline-tant | SB-222.200 | SB-218.795 | |
| Caco-2 permeability (cm-s-1) | -5.12 | -4.98 | -5.16 | -4.55 | -4.74 | -4.95 |
| Pgp substrate (probability) | 0.06 | 0.40 | 0.09 | 0.13 | 0.10 | 0.06 |
Caco-2 permeability values of all six compounds were reported to be >-5.15 cm-s-1 suggesting optimal absorption and membrane permeability [40]. Assessing Caco-2 permeability has been widely used to predict drug absorption with considerable accuracy [42]. In silico analysis carried out in the present study indicated that all six NK3R antagonists are likely GI absorbable.
P-glycoprotein acts by extruding absorbed xenobiotics from cells [43], hence assessing substrate likeliness is of great importance. No compound exhibited a probability >50%, yet Osanetant showed a considerable 40% probability of being a P-glycoprotein substrate.
Bioavailability estimation was carried out as stated in [44] with an accuracy of 70%. Cut-off points of 20% and 30% when orally administered were selected for this study. Results are reported as probability values of achieving a bioavailability (F) greater than 20% or 30% (table 3).
Table 3 Bioavailability prediction for NK3R antagonists.

F >20%: Bioavailability>20%; F >30%: Bioavailability>30%.
When analyzing the probability of reaching a bioavailability greater than 20%, suspended trial drugs and human-tested drugs exhibited an oddly same value of 0.73, still, the reported probability was over the 0.50 threshold of ADMETlab. While SB-218.795 scored the lowest, SB-222.200 showed the highest probability when compared to all previously mentioned groups.
Regarding >30% bioavailability, the highest value was exhibited by Fezolinetant, whereas the lowest was for Osanetant. Besides Osanetant, all the compounds were above the 0.50 threshold. In concordance with our results, suspended trials for Osanetant reported a slight low bioavailability when compared to novel drugs under current testing [8].
Excretion evaluation was carried out by assessing half-life and clearance [45] with an accuracy of 85% and 88%, respectively. Results of the evaluated parameters for each compound are shown in table 4.
Table 4 Elimination. Clearance and half-life of NK3R antagonists.
| Parameter | Suspended trial | Human tested | Animal tested | |||
|---|---|---|---|---|---|---|
| Talnetant | Osanetant | Pavinetant | Fezolinetant | SB-222.200 | SB-218.795 | |
| Half-life (h) | 2.18 | 1.91 | 1.81 | 1.41 | 2.10 | 2.07 |
| Clearance (cm3-min-1-kg-1) | 2.14 | 1.02 | 1.37 | 1.54 | 1.92 | 1.67 |
The predicted half-life for NK3R antagonists was reported to be within a range of 1.41 to 2.18 hours. Obtained values were over 0.50 and below 3.00 hours suggesting druglike features, however, they still exhibited a moderately low half-life [22]. Reported low half-life values indicate that an optimal therapeutic effect is achieved only with an increased dosing regimen [46], hence these results show that it is advisable to carefully evaluate dosing when analyzing these compounds as therapeutic agents. Interestingly, animal-tested drugs (SB-222.200 and SB-218.795) were reported to have higher halflife when compared to the other two groups, and on the contrary, human-tested drugs exhibited the lowest half-life values of all six compounds.
Clearance results were all <5 cm3-min-1-kg-1, suggesting that the clearance rate for these compounds is rather low [22]. Talnetant exhibited the highest clearance (2.14 cm3-min-1-kg-1), being the only compound that reported >2 cm3-min-1-kg-1. These results support Talnetant clinical trial suspension based on the poor phar-macokinetic profile as proposed by Malherbe et al. [8]. In contrast, Pavinetant and Fezolinetant seem to exhibit lower clearance and a considerably different pharmacokinetic profile in terms of excretion. Animal-tested drugs, on the other hand, reported intriguingly high values of both clearance and half-life.
Metabolism evaluation was carried out as detailed in [47]. Results are reported as the probability of inhibition or substrate interaction with CYP450 enzymes (table 5).
Table 5 CYP450 predicted inhibition and substrate-like properties.
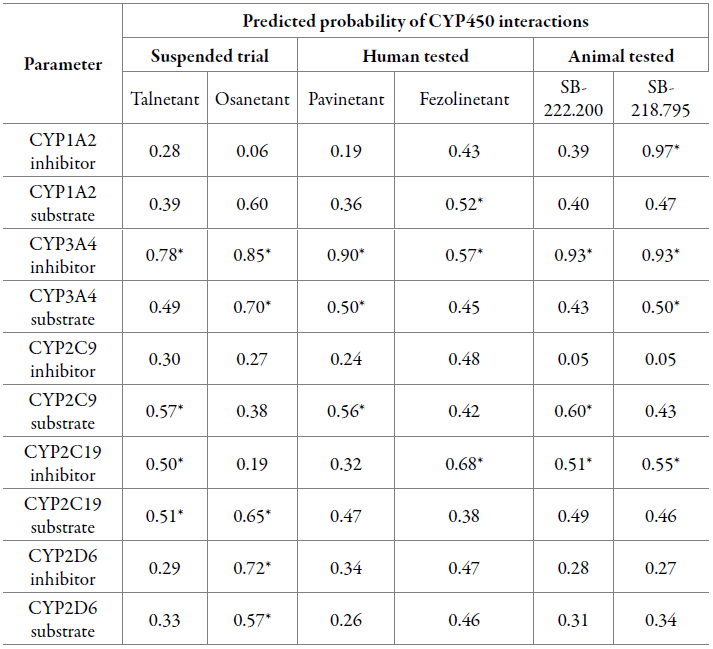
*Values that label the compound as a feasible inhibitor/substrate.
Given ADMETlab categorization, feasible interactions are likely to occur when the probability is higher than 0.50, hence every compound showed at least one interaction with a CYP450 enzyme. Most importantly, all six NK3R antagonists were reported to have induced toxicity based on their predicted CYP3A4 inhibition, still, Fezolinetant exhibited a much lower probability for the occurrence of this particular interaction. Osanetant showed the highest number of interactions with cytochromes, for which two out of five were inhibitions. On the other hand, Talnetant and SB-218.795 reported four interactions with two and three possible inhibitions, respectively. Pavinetant, Fezolinetant and SB-222.200 showed three interactions including both inhibitions and substrate-enzyme. For all three groups, the mean probability values of CYP450 interactions are shown in figure 6.

Figure 6 Probability of CYP450 interactions. Mean probability values for inhibition and substrate properties ± SEM. SEM: standard error of the mean; CYP450: cytochrome P450.
The statistical analysis reported no significant difference (p>0.05) in terms of probability, yet clear differences were evidenced. Talnetant and Osanetant exhibited a higher number of CYP450 substrate interactions, while animal-tested drugs (SB-222.200 and SB-218.795) showed greater CYP450 inhibitions. Human-tested drugs mean interacting probability lies between both aforementioned groups.
The evaluation of cytochrome interactions with drug-like compounds is crucial. As CYP450 enzymes are tightly involved in xenobiotic metabolism, the interacting probability of Talnetant and Osanetant with CYP450 indicates a higher catabolic rate leading to poor efficacy as therapeutic agents [48], most importantly, lower substrate-enzyme interactions were reported for novel NK3R antagonists. Inhibition, on the other hand, was greater for animal-tested drugs, thus these compounds are more likely to produce drug-induced hepatotoxicity when compared to human-tested drugs [49].
Toxicity profiles for NK3R antagonists were determined by the estimated probability of hERG blockage [50], human hepatotoxicity (H-HT) [51], and drug-induced liver injury (DILI) [52] with an accuracy of 84%, 69% and 84%, respectively. Results are shown in table 6.
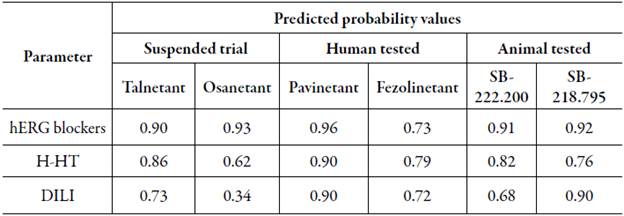
hERG blockage was found to be considerably elevated, with five compounds reporting a probability >90%, nonetheless, Fezolinetant hERG blockage probability was significantly low in comparison. H-HT probability was found to be within a range of 0.62-0.90, with Pavinetant scoring the highest and, oddly, Osanetant the lowest. Assessment of DILI probability was also within a range of 0.34-0.90, reporting the lowest for Osanetant and the highest for Pavinetant and SB-218.795. Data show that Pavinetant exhibits the greatest toxicity profile based on in silico evaluation, and in concordance with these results, clinical trial outcomes showed higher toxicity for Pavinetant as a result of an increase in subjects serum transaminases, which was not evidenced with Fezolinetant administration [12].
CONCLUSION
In silico analysis revealed a physicochemical and pharmacokinetic profile that enabled a non-previously carried out comparison between the novel and suspended NK3R antagonists. Given Talnetant and Osanetant poor outcomes in clinical trials, and as expected in this study, results show that drugs currently undergoing clinical trials (Pavinetant & Fezolinetant) and animal-tested drugs (SB-222.200 & SB-218.795) exhibited higher compliance with drug-likeness rules. When compared to trial-suspended drugs, human-tested compounds reported better physicochemical features based on molecular descriptors calculation. Because of these facts, results suggest more suitable drug-like properties for novel Fezolinetant and Pavinetant, and thus, a likely enhanced activity as NK3R antagonists. ADME/T evaluation showed considerable disparities between groups, yet no significant difference was reported in terms of absorption, distribution, metabolism, excretion, and toxicity. Results derived from this study served to elucidate crucial pharmacological and chemical traits of novel drug-like compounds currently under study for hot flashes therapy.

