











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
Permalink
La enfermedad de Pompe (EP) es un trastorno metabólico hereditario en el que la forma infantil suele ser severa y homogénea, con síndrome de bebé flácido, hipertrofia cardiaca e insuficiencia respiratoria. La presentación clínica de la enfermedad de Pompe de inicio tardío (LOPD) es más benigna y marcadamente heterogénea, con compromiso respiratorio y musculoesquelético variable, lo que hace más difícil un diagnóstico temprano (véase artículo sobre manifestaciones clínicas). En este artículo exploraremos las tecnologías y enfoques disponibles para el proceso de diagnóstico de la enfermedad.
El análisis de datos del registro de Pompe entre 1.079 pacientes muestra un retraso en el diagnóstico de la enfermedad 1. En la forma infantil, la media de retraso en el diagnóstico es de 1,4 meses en recién nacidos con miocardiopatía y otros síntomas, en los primeros 12 meses de vida. En pacientes con inicio de la sintomatología después de los 12 años, la media de retraso es de 6 años, mientras que en los pacientes con inicio de síntomas durante los primeros 12 meses de vida, sin miocardiopatía, se informó un retraso más largo de 12,6 años para el diagnóstico de la enfermedad. Por último, se observó un retraso similar en pacientes con EP que desarrollan síntomas entre los 12 meses y los 12 años; de ahí la importancia de sospechar y diagnosticar la enfermedad lo antes posible 1-3.
Puesto que la forma de presentación inicial puede ser muy variable, desde signos y síntomas leves como mialgia, fatiga, hiperCKemia aislada, intolerancia al ejercicio, calambres y debilidad muscular, hasta síntomas de insuficiencia respiratoria, es importante conocer las principales herramientas que se utilizan en la actualidad en el diagnóstico de pacientes con sospecha de enfermedad de Pompe (tabla 1).
Tabla 1 Métodos diagnósticos de laboratorio actuales
| Método | Tiempo | Invasivo | Falso - | Falso + | Pros | Cons |
|---|---|---|---|---|---|---|
| Bioquímica | ||||||
| Gota seca en papel de filtro | 2-4 días | No | Sí | Sí | Económico, rápido, fácil de realizar, útil como herramienta de tamizaje | Confirmación bioquímica o genética |
| Fibroblastos | 4-6 semanas | Sí (mínimo) | No | No | Factible, resultados precisos | Largo periodo de espera |
| Leucocitos | 2-4 días | No | No | No | Fácil de realizar | No siempre es preciso |
| Músculo | 2-4 días | Sí | No | No | Factible, resultados precisos, tiempo de espera moderado | Solo en centros esquelético especializados, usualmente en adultos |
| Morfología | ||||||
| Frotis de | Inmediato | No | No | Sí | Económico, rápido, sugestivo | Pocos estudios, requiere confirmación |
| Morfología muscular | 2-4 semanas | Sí | Sí | Sí | Si positivo, altamente sugestivo | Solo en centros especializados, requiere confirmación |
| Análisis genético | ||||||
| Secuenciación GAA | Algunas semanas | No | No | No | Altamente fiable si hay dos mutaciones patogénicas | Si es inconcluso (solo una mutación), se requiere confirmación bioquímica |
Fuente: modificado de 4.
Herramientas diagnósticas
Análisis no específicos
Los exámenes de laboratorio muestran niveles elevados de enzimas musculares como CK, LDH, AST y ALT. La hiperCKemia a menudo no es más de cinco veces de los rangos de referencia; la elevación suele ser mayor en pacientes jóvenes en comparación con aquellos con una enfermedad de mayor duración, e incluso puede ser normal en pacientes con enfermedad de inicio tardío 4.
Un potencial biomarcador de enfermedades por almacenamiento de glucógeno (GSD) es el tetrasacárido 6-α D-glucopiranosil-maltotriosa (Glc4), debido a que la excreción urinaria de Glc4 aumenta en diferentes condiciones clínicas asociadas con un mayor recambio o almacenamiento de glucógeno. Aunque esta prueba es sensible y precisa para un diagnóstico presuntivo, no es capaz de diferenciar los tipos de GSD. Este ensayo debe usarse en combinación con los análisis genéticos y enzimáticos estándar para confirmar el diagnóstico de EP 5.
Estudios neurofisiológicos
En la LOPD, en la cual la afectación del músculo esquelético es una característica destacada, es útil hacer estudios electrofisiológicos para considerar el diagnóstico diferencial con otros trastornos musculares, enfermedad motoneuronal y descartar patología de nervio.
El estudio de electromiografía (EMG) revela un patrón miopático con potenciales de fibrilación, ondas agudas, descargas complejas repetitivas y evidencia de descargas miotónicas (DM) eléctricas, sin presencia de miotonía clínica 6. Estas DM parecen más comunes en los músculos paraespinales y en el tensor de la fascia lata, lo que sugiere que estos músculos deben incluirse durante la EMG de los casos sospechosos 6,7.
Biopsia muscular
El valor diagnóstico de la biopsia muscular en pacientes con LOPD es bastante limitado, porque en diferentes grupos de músculos e incluso fibras dentro del mismo grupo de músculos, se observa una alta variabilidad.
La evaluación histológica del espécimen de la biopsia muscular para EP muestra acumulación de glucógeno y una miopatía vacuolar. Estas vacuolas con frencuenca contienen glucógeno y se tiñen con PAS (periodic acid-schifj)5 (figura 1).
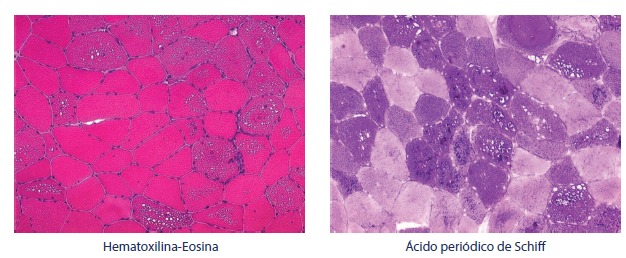
Fuente: Cortesía Prof. Edoardo Malfatti.
Figura 1 Biopsia muscular en LOPD (enfermedad de Pompe de inicio tardío). Miopatía vacuolar típica con vacuolas PAS positivas en alrededor del 70 % de los casos
Las vacuolas autofágicas son positivas para fosfatasa ácida tanto en las fibras tipo I como en las tipo II. El glucógeno puede ser visto en microscopia electrónica en los lisosomas, y también en el citoplasma; aunque esta técnica no se utiliza frecuentemente, debería usarse, si está disponible 8,9. Sin embargo, la identificación histológica de las inclusiones de lipofuscina positivas a la fosfatasa ácida se sugirió como un marcador de diagnóstico de LOPD en músculo esquelético 1.
Resonancia muscular
La resonancia magnética (RMN) de músculo puede ser útil para identificar patrones típicos de remplazo de músculo por grasa en las imágenes de T1w, lo que puede guiar al diagnóstico de la enfermedad en pacientes adultos que presentan síntomas de debilidad proximal-cintura miembro, al simular distrofias musculares 10.
Ya que hasta ahora la enfermedad de Pompe es la única miopatía hereditaria con tratamiento aprobado, la RMN proporciona la oportunidad de monitorizar cambios en la estructura muscular, en el contenido de grasa y en el glucógeno como respuesta a un tratamiento de remplazo enzimático 11.
Los estudios semicuantitativos de las secuencias T1w se han utilizado para el diagnóstico tanto en EP de inicio temprano como en el tardío. En el caso de Pompe de inicio tardío, los estudios T1w muestran un patrón de compromiso considerado característico y útil para el diagnóstico. Los músculos más comúnmente remplazados por grasa son: la lengua, el sub-escapularis, el latissimus dorsal, los paraespinales, los abdominales, el psoas, el aductor magnus, los músculos posteriores del muslo, el semimembranoso y el vasto intermedio. En cambio, los músculos rectus femoris, gracilis y sartorio no son afectados sino hasta etapas muy avanzadas. En la mayoría de los casos, es más severo el grado de remplazo graso en los miembros inferiores que en los superiores 10 (figura 2).

Fuente: cortesía Prof. Jordi Díaz-Manera.
Figura 2 Resonancia muscular. En estas imágenes de resonancia muscular observamos la infiltración grasa en los músculos aductores mayores, comparada con una resonancia normal
La resonancia de músculo es capaz de identificar remplazo graso aun en fases presintomáticas. En resumen, los músculos más afectados en la resonancia son: la lengua, los paraespinales, el abdomen y el aductor magnus. Con base en estos hallazgos, la resonancia es muy útil para diferenciar de otros trastornos neuromusculares, dado que los músculos distales generalmente están preservados en la enfermedad de Pompe, al contrario de algunas formas de distrofia muscular, tal como la distrofinopatia, la disferlinopatia y la calpinopatia, que tienen más compromiso de los músculos distales, principalmente gastrocnemius medial y soleus. Sin embargo, las sarcoglicanopatias tienen un patrón similar al visto en la EP con compromiso de músculos abdominales y paraespinales, pero a pesar de ello tienen un mayor compromiso de miembros superiores, sobre todo en los músculos bíceps, tríceps y escapularis. En cambio, el compromiso de la lengua es típico de la EP 10.
Análisis de actividad enzimática GAA
El estándar de oro para las pruebas de diagnóstico en la enfermedad de Pompe es la identificación de la actividad enzimática GAA disminuida o ausente. Esta anormalidad se ha medido en muestras de tejido, incluidos los fibroblastos de la piel, el músculo y también en sangre 5.
Análisis de actividad enzimática GAA en muestras de sangre
La actividad de la enzima GAA puede medirse con precisión en muestras de sangre como DBS (obtenida de sangre completa) y los linfocitos purificados.
Gota seca en papel filtro
Ante la sospecha de EP, la prueba más fácil y confiable es la de la gota seca en papel filtro (DBS), utilizado como primera elección para el tamizaje. Por lo general, se utilizan 2 métodos para el análisis de estas muestras: fluorometría o espectrometría de masas, ambas disponibles para el diagnóstico, aun en pruebas de tamizaje para recién nacidos. Puede producirse una prueba de DBS falso positivo, debido a una extracción y un muestreo de sangre incorrectos, como también producto de circunstancias ambientales como altas temperaturas durante el transporte 4.
El examen detallado de la actividad de la enzima en sangre puede revelar la presencia de otras alfa glucosidasas como la maltasa glucoamilasa (MGA), la cual puede enmascarar la deficiencia de GAA. Esta interferencia de la enzima MGA fue la mayor limitación en ensayos previos, que conducían a resultados falsos negativos. El uso de acarbosa para la inhibición competitiva de la MGA en los nuevos ensayos elimina este problema 5.
Linfocitos purificados
La prueba de linfocitos purificados se utiliza por la ausencia de MGA en estas células, sin embargo, este método aún podría dar algunos datos falsos negativos si la muestra está contaminada con neutrófilos 12. Por ello, se utiliza con más frecuencia la medición de la actividad de la enzima GAA en cultivo de fibroblastos de piel o en muestras de biopsia muscular.
Cultivo de fibroblastos en piel
En relación con la actividad enzimática de GAA en cultivo de fibroblastos de piel, esta muestra debe ser tomada por personal idóneo con experiencia en este método, pero además, el ensayo enzimático se realiza solo después de cultivar los fibroblastos, lo que puede tardar de 4 a 6 semanas, con un retraso significativo en el diagnóstico; por esta razón, ya no se utiliza como primera elección diagnóstica, pero sí como prueba confirmatoria luego de un DBS positivo 4.
Cultivo de linfocitos en sangre periférica
En la enfermedad de Pompe, hay una acumulación generalizada de glucógeno en todos los tejidos y también en las células sanguíneas. Hace algunos años se observó que la acumulación de glucógeno estaba presente en los linfocitos, en los cuales se observaba vacuolización en el frotis de sangre periférica (BSE) 13.
Estudios recientes demostraron que la evaluación de linfocitos vacuolados en la BSE es una herramienta confiable en el diagnóstico diferencial de miopatías autofágicas 14. Sin embargo, los resultados de la BSE deben confirmarse mediante la detección de la deficiencia enzimática o el análisis genético.
Actividad enzimática GAA en muestras de tejido
GAA muscular
Aunque es más invasiva que las muestras de piel, la biopsia de tejido muscular nos da información en cuanto a los hallazgos de patología, y además va a determinar la actividad residual de la enzima 15. Se debe tener especial precaución en la toma de la muestra, dado que una parte de la biopsia se envía a histología y la otra parte debe ser apropiadamente congelada para la medición de la actividad de la enzima 12.
Las investigaciones han demostrado que el test de GAA muscular es una prueba fiable para detectar la deficiencia de GAA y que los grados de deficiencia de la enzima determinan el curso de la enfermedad. Los valores superiores al 35 % de los rangos de referencia sugieren enfermedad de Pompe, mientras que una reducción de la actividad GAA en fibroblastos o músculo por debajo del 30 % es consistente con el diagnóstico de LOPD 16.
Finalmente, hay que enfatizar que ante la sospecha de enfermedad de Pompe, tanto en niños como en adultos, se debe hacer la prueba de secuenciación del gen de la enzima de la alfa glucosidasa ácida para confirmar el diagnóstico. La figura 3 muestra el algoritmo sugerido para el diagnóstico de la EP.

Fuente: tomado y adaptado de 7.
Figura 3 Algoritmo diagnóstico de la enfermedad de Pompe EMG: electromiografia, CPK: creatinfosfoquinasa, CVF: capacidad vital forzada, GAA: alfa-glucosidasa ácida.
Diagnóstico molecular en la enfermedad de Pompe
Puesto que las variantes afectan directamente el cuadro clínico, y el diagnóstico temprano es esencial para prevenir y reducir el daño orgánico asociado con la progresión de la enfermedad 17, el consenso europeo de EP establece como estándar de oro para el diagnóstico, la combinación del ensayo enzimático con secuenciación genética 18.
La EP es un trastorno autosómico recesivo, lo que requiere la existencia de una variante patogénica en ambas copias del gen GAA para que se manifieste la enfermedad 19,20,21. El gen GAA se localiza en el brazo largo del cromosoma 17 (17q25.2- q25.3) 22, tiene una longitud de w28 kb y contiene 20 exones. El primer de estos no codificante, contiene secuencias no traducidas de 5' y está separado del segundo exón por un intrón de w2.7-kb. El primer codón de inicio, ATG, se encuentra 32 nucleótidos corriente abajo del comienzo del exón 2, donde se inicia la codificación de una proteína de 952 aminoácidos, con un peso molecular de 110 -kDa 22,23,24.
Si se considera que el gen GAA es responsable de la producción de proteína α - glucosidasa ácida, la aparición de variantes patogénicas (VP) a lo largo de este gen se verá directamente reflejada en los valores enzimáticos, lo que es evidencia de una deficiencia de la enzima GAA y una actividad enzimática anormal 25, influye en la gravedad de las manifestaciones y contribuye a la variación observada en la edad de presentación y en la tasa de progresión 25,26,27,28,29,30.
Gracias a la implementación del screening neonatal para EP en algunos países y al registro de las variantes genéticas (http://www.pompevariantdatabase.nl/) 31, cada día se cuenta con más información sobre las variantes en el gen GAA. En la actualidad, se han informado 911 variantes genéticas en el gen GAA, de las cuales el 71 % se asocia con la presentación de la enfermedad; en un 52 % de estas se ha logrado establecer un fenotipo clínico específico; el 29 % restante se consideran variantes de significado clínico incierto (VUS) 32. Estas corresponden a mutaciones puntuales o a pequeñas y grandes deleciones e inserciones, donde se puede afectar la funcionalidad y la estabilidad de la proteína, ya sea por modificaciones en el proceso de corte y empalme (splicing), que ocasiona la alteración en la transcripción del ARN y en la posterior síntesis de proteínas, o genera modificaciones postraduccionales, incluida la glicosilación, el tráfico lisosómico y la naturaleza proteolítica de GAA 33,34.
De conformidad con lo mencionado anteriormente sobre el gen GAA identificado como gen altamente polimórfico, con varias variantes neutrales, los estudios moleculares se convirtieron en una herramienta común para confirmar el diagnóstico de EP e identificar las variantes asociadas a la enfermedad 32. La secuenciación de Sanger y la secuenciación de nueva generación por secuenciación de nueva generación (NGS) se vuelven el método más eficaz para la identificación de variantes de un solo nucleótido en el gen GAA 35.
A partir de que la EP es un trastorno autosómico recesivo, si mediante la secuenciación no se identificó una VP o una variante probablemente patogénica (VPP) en homocigosis, o dos variantes diferentes en cada uno de los alelos -como en la heterocigosis compuesta, donde se requiere además la confirmación mediante un estudio de segregación en familiares 36,37 con el fin de demostrar que las dos variantes están en alelos diferentes-, el paso por seguir es evaluar ganancias o pérdidas de información génica en el gen GAA, para lo cual las técnicas indicadas son en primera instancia el estudio de amplificación de sonda dependiente de ligadura múltiple (MLPA) o la realización de un CGH array 35. Cuando los procedimientos estándar son insuficientes para validar el diagnóstico de EP, se pueden hacer análisis de diagnóstico molecular extendidos, como un ensayo de empalme genérico, análisis de minigén, análisis de matriz de polimorfismos de un solo nucleótido (SNP) y secuenciación de Sanger dirigida 38.
El espectro mutacional de GAA con estás técnicas de diagnóstico molecular es muy heterogéneo, la mayoría de las variantes genéticas se han reportado como exclusivas de una familia o se encuentran en una pequeña población 39,40. Otras variantes son más frecuentes o comunes y se pueden observar en diferentes grupos étnicos: en afroamericanos la variante más frecuente es p.R854X 27,41,42, en población China es la variante p.D645E A 39, en los Países Bajos del 525T y c.925G> A, y en los pacientes taiwaneses la VP más común es c.1935C> A 43,44. El defecto más frecuente en población caucásica con enfermedad de inicio tardío es la variante intrónica, que ocasiona una alteración en el splicing y conduce a la omisión del exón 2 (c.-32-13T> G IVS1) 45, lo que genera niveles bajos (10-20 %) de los valores enzimáticos normales 46,47,48. Además, se ha demostrado en un subconjunto de estos pacientes que el alelo mutante IVS1 contiene un modificador genético (c510C> T; una variante sinónima) que reduce aún más la cantidad de enzima activa 49. En Colombia no existen datos específicos sobre la prevalencia de la EP. Hasta el 2013 se informaron 24 casos, así como las mutaciones p.E176fsx45, p.L355P,p.W746R, p.G828_N882del y p.R854X 50,51.
Aunque no hay una correlación estricta entre el genotipo y el fenotipo, la presencia de mutaciones sin sentido en ambos alelos da como resultado una proteína truncada que se asocia con la EP clásica de inicio infantil (IOPD) 52. Se ha determinado además que la asociación de dos variantes: c.1726G> A y c.2065G> A, a menudo presentes en cis, da lugar a una pseudodeficiencia de GAA. El c.1726G> A afecta tanto a la cantidad de GAA como a su actividad catalítica, mientras que c.2065G> A reduce ligeramente la funcionalidad de GAA 26,53,54,55.
Otro aspecto importante de los hallazgos moleculares reportados en la EP es el relacionado con el estado del material inmunológico de reacción cruzada (CRIM), que funciona como un predictor de la respuesta a la terapia de reemplazo enzimático (TRE) 56. En los pacientes que se identifican con un estado CRIM negativo (CRIM-), no se sintetiza proteína GAA debido a la presencia de alelos GAA nulos. Por tanto, la rhGAA se reconoce como una proteína extraña por el sistema inmunológico en estos pacientes, que resulta en el desarrollo de niveles altos de títulos de anticuerpos neutralizantes, lo cual hace que la terapia sea ineficaz 56,57.
Un estudio llevado a cabo en 243 niños de diversas etnias, con diagnóstico de EP de inicio infantil, mostró que 61 (25,1 %) eran CRIM- 56. Las mutaciones identificadas con mayor frecuencia fueron p. Arg854X y c.525delT. La mayoría de los pacientes eran homocigotos o heterocigotos, compuestos para mutaciones sin sentido o cambio de marco de lectura, que daban como resultado codones de parada prematuros o deleciones de múltiples exones. No se identificaron mutaciones sin sentido en los pacientes CRIM. Sin embargo, un paciente CRIM- fue homocigoto para una mutación puntual que abolió la metionina iniciadora (c.1A> G; p. Met1?). Por el contrario, la mayoría de los pacientes CRIM-positivos (CRIM +) tenían una o dos mutaciones de deleción sin sentido o en marco de lectura que predecían la síntesis de alguna proteína GAA. Ninguno de los pacientes CRIM + presentó la misma combinación de mutaciones de los pacientes CRIM - 56.
Asesoramiento genético y diagnóstico prenatal
La enfermedad por acumulación de glucógeno tipo II es una enfermedad hereditaria autosómica recesiva. Los padres del probando suelen ser portadores y el riesgo de recurrencia es del 25 %.
Todos los casos y sus familiares deben recibir asesoramiento genético; el diagnóstico prenatal de fetos de alto riesgo y el diagnóstico genético previo al implante debe aplicarse a las familias en las que se ha identificado la variante patógena.
Debido a la presencia de alelos pseudodefectuosos (C.1726G> A o c.2065G> A), el análisis genético debe ser la primera opción para el diagnóstico prenatal, en tanto que la detección de variantes patogénicas claras en la familia, la comprensión del estado portador de los alelos defectuosos falsos y la combinación con la actividad de la enzima GAA son obligatorias.
Si en el paciente no se encuentran dos variantes patogénicas claras, puede elegirse la medición de la actividad enzimática GAA para proporcionar un diagnóstico prenatal, comprender el estado de los alelos de pseudodefecto e interpretar los resultados con cuidado 57.
El diagnóstico prenatal de rutina se suele llevar a cabo mediante biopsia de las vellosidades coriónicas entre las 11 y las 13 semanas de gestación, o con amniocentesis entre las 17 y las 22 semanas de gestación, para obtener células fetales 57. Los resultados del diagnóstico prenatal no pueden predecir la edad de aparición, el curso clínico o la gravedad de los fetos portadores de genes variantes patógenos con enfermedad de Pompe después del nacimiento.
Tamizaje neonatal
El diagnóstico precoz, especialmente el diagnóstico presintomático y la terapia de reemplazo enzimático (TRE) precoz pueden mejorar significativamente el pronóstico de la enfermedad de acumulación de glucógeno infantil tipo II.
El tamizaje de recién nacidos utiliza principalmente espectrometría de masas en tándem o método de fluorescencia para detectar la actividad enzimática GAA de muestras en papel filtro con sangre seca 57,58. Para aquellos que dan positivo en las pruebas de detección, se recomienda recolectar leucocitos o linfocitos de sangre periférica para la determinación de la actividad de la enzima GAA. Si la actividad de la enzima GAA esta disminuida, se debe realizar asesoramiento genético, evaluación clínica multidisciplinaria y análisis de mutación del gen GAA al mismo tiempo, para confirmar el diagnóstico y el subtipo.
Diagnóstico diferencial
Debido a la heterogeneidad de la presentación, la forma tardía (LOPD) tanto en niños como en adultos puede simular otros trastornos neuromusculares, y el diagnóstico diferencial incluye una amplia gama de miopatías, así como enfermedades de la neurona motora y de la unión neuromuscular.
La LOPD debe diferenciarse de las siguientes enfermedades: distrofia muscular de cinturas, distrofia muscular de Becker, síndrome escapuloperoneal, miastenia gravis, atrofia muscular espinal, polimiositis, enfermedad por acumulación de glucógeno tipo IIIa , tipo IV, tipo V y tipo VII, enfermedad de Danon y miopatía mitocondrial, entre otras. 5,58,59,60,61 (tabla 2).
Tabla 2. Diagnóstico diferencial de Pompe de inicio tardío
| Tipo de trastorno | Diagnósticos |
|---|---|
| Distrofias |
|
| Miopatías inflamatorias |
|
| Miopatias congénitas |
|
| Otras miopatías metabólicas |
|
| Trastornos de neurona motora |
|
| Trastorno de unión neuro-muscular |
|
Fuente: modificado de 7.
La IOPD, por su parte, debe diferenciarse de las siguientes enfermedades: atrofia muscular espinal infantil tipo 1, distrofia muscular congénita, fibrosis elástica endocárdica, miocarditis, miocardiopatia tipo hipertrófico primaria, hipotiroidismo congénito, enfermedad de acumulación de glucógeno tipo IIIa y tipo IV, deficiencia primaria de carnitina, enfermedad de Danon, miopatía mitocondrial y síndrome de Prader-Willi, entre otras 15,61.

