











 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
PermalinkIntroduction
Lecithin-cholesterol acyltransferase (LCAT) deficiency is a rare autosomal recessive genetic disease, with an es timated global prevalence of 1/1,000,000 inhabitants. It presents with the classic triad of diffuse corneal opacities, hemolytic anemia and kidney disease, the latter represent ing the main cause of morbidity and mortality. It begins in childhood with proteinuria, progresses to chronic kidney disease toward the fourth or fifth decade of life, and there is no effective treatment available at this time. In addition, patients who undergo kidney transplantation have been found to have a rapid recurrence of the disease in the graft 1,2. To date in Colombia and Latin America, there has been little information reported on this disease, and therefore this case contributes to the knowledge of the clinical presen tation of a rare disease, with a diagnosis which requires a high index of suspicion integrating a broad constellation of signs, symptoms and laboratory findings which are usually treated in an isolated fashion and attributed to other causes.
Case report
This was a 30-year-old Hispanic male patient who had consulted six years prior for lower extremity edema. His tests showed 24-hour urine proteinuria at 4.3 gm, albumin 2.8 gm/ dL, triglycerides 1,388 mg/dL, total cholesterol 263 mg/ dL, high density lipoproteins (HDL) 5.0 mg/dL, low density lipoproteins (LDL) 19 mg/dL, creatinine 1.2 mg/dL, proteinuria at 500 mg/dL, without hematuria, and negative tests for active infection, immune disease and neoplasms. A kidney biopsy reported focal segmental glomerulosclerosis (FSGS) interpreted as primary. He was initially treated with high-dose prednisone and mycophenolate mofetil, but when his proteinuria did not improve, he received intravenous cyclophosphamide followed by cyclosporine for three years and, finally, tacrolimus, with no response.
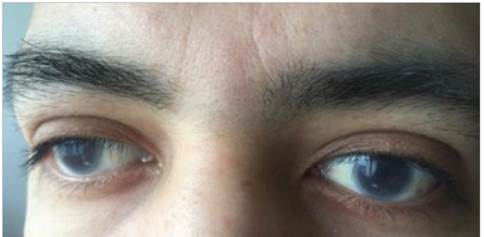
He presented to the emergency room with right upper quadrant pain associated with nausea, five episodes of vomit ing and persistent extremity edema, with no other relevant findings on the review of systems. In his family history, the patient's parents were blood relatives (cousins). He had a 22-year-old sister with dyslipidemia and corneal opacities similar to his own. His vital signs were normal, and the physical exam showed bilateral corneal opacities, with no abnormal cardiopulmonary findings; pain on palpation in the right upper quadrant with no signs of peritoneal irrita tion or palpable hepatomegaly or splenomegaly; and grade II edema in the lower extremities. The rest of the exam was within normal limits.
The laboratory tests reported the following values: creatinine 2.35 mg/dL, blood urea nitrogen 50 mg/dL, 24-hour urine proteinuria 10.2 gm, albumin 1.19 gm/dL, triglycerides 846 mg/dL, total cholesterol 194 mg/dL, LDL cholesterol 10 mg/dL, HDL cholesterol 13.9 mg/dL, hemoglobin 7.6 gm/dL, hematocrit 20.4%, and mean cor puscular volume 86 fl. The rest of the complete blood count was normal with a corrected reticulocyte count of 0.1%, a negative direct Coombs, total iron 65.9 u/dL, transferrin 125 mg/dL, and a transferrin saturation index of 36%. Serum protein electrophoresis and a bone marrow biopsy ruled out paraproteinemia. The studies for viral hepatitis, syphilis and human immunodeficiency virus were negative, as were the antinuclear antibodies. Complement C4 was normal and C3 was consumed (54.9 mg/dL). An alpha-galactosidase activ ity assay ruled out Fabry disease. An abdominal ultrasound showed a hydrocholecyst and 15.9 cm splenomegaly; both kidneys were of normal size.
The findings of corneal opacities, anemia, dyslipidemia with very low HDL, kidney failure and severe nephrotic syndrome refractory to treatment with multiple immunosup pressants oriented toward a possible diagnosis of complete lecithin-cholesterol acyltransferase deficiency. Complete sequencing of the LCAT gene was requested, and the study identified the c.368G > C homozygous variant which cor responds to the exchange of a guanine for a cytosine in position 368 of the coding DNA. This variant causes a missense change of an arginine for a proline in amino acid 123. Later, a kidney biopsy showed thickening of the capillary basement membrane and mesangial matrix due to multiple lipid vacuoles. The molecular study results together with the kidney biopsy findings confirmed the diagnosis.
Discussion
Lecithin-cholesterol acyltransferase deficiency is a rare autosomal recessive disease, with an estimated global prevalence of 1/1,000,000 inhabitants. To date, 60 isolated cases have been identified, along with 70 families with complete or partial LCAT deficiency. The disease is caused by a mutation of the gene coding for the LCAT enzyme, located in the 16q22.1 chromosome region. This enzyme plays an essential role in the metabolic pathway for reverse cholesterol transport, changing immature discoid HDL into mature spherical HDL through esterification of free cholesterol into a cholesterol ester. The mutation may be homozygous, as in this case, and cause complete enzyme deficiency (Norum disease) or it may be heterozygous, causing partial enzyme deficiency, in which case it has a different presentation and is known as fish-eye disease 1.
The Human Gene Mutation Database (HGMD) has de scribed 102 functionally relevant variants of the LCAT gene and 77 point mutations, including the mutation reported in this case, a homozygous c.368G > C mutation causing a p.Arg123Pro missense change, which has a very low popu lation frequency (< 0,0001 gnomAD), and is not reported in the available Latin American literature 1.
Clinically, the lipoprotein metabolism abnormality manifests with markedly low HDL (< 10 mg/dL) and a decreased LDL due to accelerated catabolism, while the VLDL, free cholesterol and triglycerides are elevated 2. In this case, the patient had been diagnosed with severe hypertriglyceridemia at age 22 and was treated with fibrates. He also had markedly low HDL and LDL, abnormalities which, together with the diffuse corneal opacities (which are the earliest and most well-known sign of the disease and which were not initially considered to be relevant), are characteristic of LCAT deficiency 2.
Kidney involvement manifested in adolescence as a nephrotic syndrome. He was initially diagnosed with focal segmental glomerulosclerosis due to findings on the first kidney biopsy, a study in which the typical histopathological characteristics of LCAT deficiency were not evident, which include thickening or duplication of the glomerular basement membrane on light microscopy, with significant mesangial proliferation, and lipid deposits on electron microscopy, consisting of small, dark, irregular and electro-lucent granular particles located mainly in the subepithelial, subendothelial, intramembranous and mesangial spaces 3,4. The absence of these findings probably contributed to a delay in the diagnosis, since it was interpreted as a primary glomerular disease, which is why he received multiple immunosuppressant treatment regimens without achieving disease remission.
In the reported literature on nephrotic syndrome due to familial LCAT deficiency, the diagnosis was made based on the typical characteristics on kidney biopsy and was subsequently confirmed through genetic sequencing. Although FSGS is not recognized in any article as an initial glomerular lesion pattern, in 1983, Moorehead et al. reported the case of a nine-year-old patient with partial LCAT deficiency, and nephrotic syndrome and FSGS on the kidney biopsy with no lipid deposits. In that case, the diagnosis was made through direct measurement of enzyme activity 5,6. An FSGS pattern in LCAT deficiency is a late finding in the course of the disease and secondary to progressive accumulation of lipid-containing, vacuolated foam cells in the mesangium and glomerular basement membrane which cause inflammation and fibrosis 7,8.
In addition to causing damage from accumulation in the kidney, lipoprotein x [Lp(x)], an abnormal lipoprotein rich in free cholesterol and phospholipids, has proven to be cy-totoxic and proinflammatory, stimulating monocyte infiltra tion through monocyte chemoattractant protein-1 (MCP-1) expression and increased nuclear factor kappa B (NF-kB) in the mesangial cells, causing an inflammatory response which plays a significant role in glomerulosclerosis 9.
The moderate, normocytic, normochromic anemia was not explained by the kidney disease and is attributed to free cholesterol and phosphatidylcholine deposits in red blood cell membranes, which shortens their average lifespan and causes hemolytic anemia that is generally low-grade and may go unnoticed. The anemia was associated with sple nomegaly caused by hyperplasia of the reticuloendothelial system resulting from the increased destruction of defective erythrocytes 10. It is notable that the bone marrow tests were normal.
To date, there is no specific treatment available. Most pa tients require renal support therapy and those able to receive a transplant have reported recurrence of the disease in the graft 7. A recombinant human LCAT (rhLCAT) enzyme is being developed for treating complete enzyme deficiency. The first studies have shown lipid profile normalization, anemia reversal, improved kidney function and transforma tion of Lp-X into HDL-like particles 7.
Conclusion
A high index of suspicion in interpreting clinical mani festations and diagnostic tests, including kidney biopsies, is necessary for diagnosing LCAT deficiency. The diagnosis should always be confirmed with sequencing of the gene coding for LCAT. Familial genetic counseling is important to determine measures aimed at decreasing the risk of kid ney disease progression, and avoiding unnecessary tests and treatment which, due to the disease's pathophysiology, prove to be ineffective and potentially toxic.

