











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
PermalinkSeñor editor, se examinó a una mujer de 28 años, originaria de Miahuatlán de Porfirio Díaz Oaxaca, México, remitida para descartar esclerosis lateral amiotrófica (ELA) y miopatía mitocondrial por la unidad de rehabilitación básica. La paciente presentaba disfagia, debilidad muscular, calambres, fatiga e intolerancia al ejercicio, síntomas progresivos desde hace 2 años. Los familiares que la acompañaban refirieron que en ocasiones presentaba llanto sin causa alguna, así como odinofagia al pasar líquidos y comida sólida desde los 13 años, lo que había progresado a disfagia.
A la paciente se le detectó sordera sensorial derecha a los tres años de edad y diabetes mellitus un año atrás, la cual se manejó con metformina 500mg cada 8 horas. No se le documentó historia de crisis convulsiva o actividad epiléptica hasta la fecha. A la exploración física no se encontró oftalmoplejía externa ni ptosis palpebral, pero sí hiperreflexia de miembros pélvicos y atrofia de los músculos de miembros torácicos muy marcada en las eminencias tenar e hipotenar. En la tomografía de cráneo contrastada no se encontraron infartos fantasmas, conocidos como stroke-like, pero la resonancia magnética simple de la columna vertebral mostró atrofia de las astas anteriores. En la historia familiar no se encontró ningún otro miembro afectado.
La exploración clínica y neuroimagenológica que se integra mediante los criterios del Escorial permitió sugerir la categoría diagnóstica de sospecha de ELA esporádica (ELAE). La paciente presentaba odinofagia y disfagia asociadas, hallazgos relacionados con la disfunción bulbar. A su ingreso a la unidad de rehabilitación, a la paciente se le solicitó una biometría hemática completa, la cual resultó dentro de los parámetros normales, y una cuantificación de ácido láctico con resultado de 8mmol/L.
Con base en estos hallazgos clínicos, se realizó un tamizaje molecular y se buscó la amplificación de los repetidos CAG en el intrón 1 del gen ATXN2 y mutaciones en SOD1 (ambos responsables de ELA13 y ELA1, respectivamente), así como la detección de mutaciones en CYTB (responsable de miopatía mitocondrial con fibromialgia). Para la detección de repetidos (CAG)n, se usaron los iniciadores PCR-Hot star, cag-F 5'-GGGCCCCTCACCATGTCG-3' y cag-R 5'-CGGGCTTGCGGACATTGG-3' (Sigma Aldrich) con las condiciones de amplificación antes reportadas 2.
Para SOD1 se flanquearon los cinco exones con los iniciadores que se muestran en la Tabla 1.

Las condiciones de amplificación fueron 95°4' (92°30", 60°25",72°25")25ciclos.
Para CYTB se utilizaron los iniciadores 5'-ATACTCCTCAATAGCCATCTC-3' y 5'-CATCATGCGGAGATGTTTGAT-3' (SigmaAldrich); las condiciones de amplificación fueron 95°4' (92° 1min, 65° 1min, 60° 30")30ciclos, extensión final 60°5"min.
En el caso del gen ATXN2 se detectaron los repetidos CAG por electroforesis en poliacrilamida al 12%. Los productos de PCR punto final de SOD1 y CYTB fueron secuenciados.
Los resultados de este estudio revelaron que la paciente es portadora de un genotipo 22/22 normal para ATXN2, negativo para mutaciones en los cinco exones del gen SOD1 y positivo para la variante G>T nt14770 en CYTB en estado heteroplásmico (Figura 1).
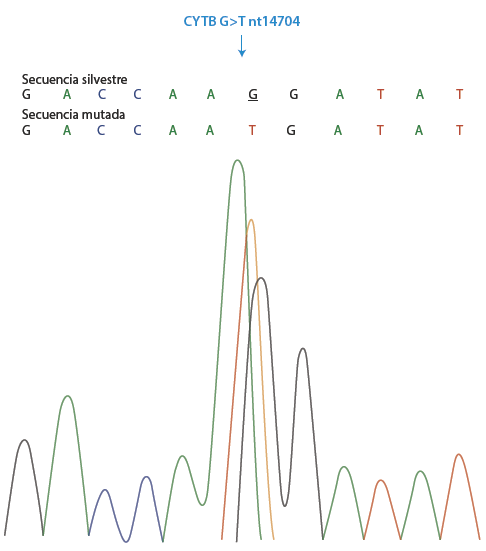
La asociación del ELA con mutaciones en genes mitocondriales es posible, ya que el estrés oxidativo está relacionado con la biogénesis, la disfunción del metabolismo mitocondrial y el control de la producción de radicales superóxido (EROS) por SOD1 y 2 4. En futuros estudios de epidemiología de ELA se deben buscar mutaciones o polimorfismos en genes mitocondriales como factores etiológicos, tal como se realizó en el presente caso, donde se exploró el gen CYTB por el fenotipo similar a fibromialgia con diabetes y sordera.
La asociación de diabetes con ELA puede explicarse por la mutación en el gen CYTB, lo que es posible ya que las mutaciones en el ADNmt se relacionan con miopatías, las cuales cursan con esta enfermedad 5. Es obligatorio descartar mutaciones en SOD, las cuales son las responsables de la mayor parte de casos con ELA familiar; en el presente caso se descartaron y era lo esperado porque se trata de un caso esporádico 6. La expansión CAG en el gen ATXN2 entre el rango de repetidos de 27-33 ha sido implicada en el desarrollo de ELA y diabetes mellitus; este estado de permutación fue descartado por estudio molecular 2,7.
En casos de ELAE, debe descartarse la expansión anormal del VTNR (GGGGCC)n en el gen C9orf72, ya que puede explicar hasta un 30% de los casos de ELA 8. En el presente caso no se realizó detección molecular de este VNTR, ya que la paciente presentaba datos clínicos que orientaban a una enfermedad de sobreposición tanto de ELA como de una miopatía mitocondrial. Los hallazgos motivaron a buscar mutaciones en genes mitocondriales que pudieran explicar la diabetes, la intolerancia al ejercicio y la fibromialgia, condiciones asociadas al gen CYTB 1.
Se ha reportado un cuadro muy parecido en el síndrome de Wolfram, el cual presenta diabetes insípida, diabetes mellitus de inicio temprano, sordera, atrofia óptica, convulsiones, retardo mental, anemia megaloblástica, leucopenia y trombocitopenia, relacionadas todas con una gran deleción de 7.6kb en estado heteroplásmico que va del nucleótido 6466 al 14134; este último locus incluye región codificante del extremo carboxilo del gen CYTB 9. En la paciente fue descartado este síndrome por el cuadro clínico, ya que la paciente no presentó acidosis láctica, alteraciones hematológicas, retardo mental, epilepsia, polidipsia, poliuria, ni infecciones repetitivas, lo cual se corroboró con la secuenciación ya que no se comprobó deleción del locus mitocondrial.
Se concluyó que la paciente analizada en el presente estudio es portadora de sospecha de ELAE asociada con la mutación G>T nt14770 en el gen CYTB.

