











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
PermalinkIntroducción
La acuicultura mundial muestra un incremento anual sostenido, según los reportes de la Organización de las Naciones Unidas para la Alimentación y la Agricultura [1]. La magnitud de extracción de productos provenientes de la pesca es oscilante, con épocas de mayor o menor biomasa marina extraída, en tanto la producción proveniente de la acuicultura es más predecible, puesto que es controlada y el proceso de cría y su rendimiento se pueden modificar a voluntad [2]. En el Perú, las exportaciones acuícolas crecieron un 25,3% en el periodo enero-octubre de 2021, hasta alcanzar 338 millones de dólares estadounidenses y 43.691 toneladas [3].
Sin embargo, los productos acuícolas pueden contener residuos o contaminantes químicos, tales como residuos de medicamentos veterinarios, resultantes del uso intencionado (administración de fármacos para contrarrestar enfermedades) o no intencionado (contaminación ambiental), lo que genera un problema de salud pública y al mismo tiempo pone en riesgo la comercialización de los productos acuícolas, sobre todo en el mercado internacional [3], [4].
Medicamentos veterinarios tales como antibióticos y antiparasitarios cumplen un rol importante en la acuicultura para el tratamiento y prevención de enfermedades transmisibles. Sin embargo, cuando su uso es indiscriminado, cuando no se cumplen los periodos de retiro y las dosis establecidas, o cuando se utilizan sustancias no autorizadas, se pueden originar residuos de estas sustancias en los tejidos comestibles, que pueden causar efectos adversos en la salud humana, desde leves reacciones alérgicas hasta resistencia bacteriana y graves efectos cancerígenos o teratógenos [5], [6]. Por ello, las agencias gubernamentales de inocuidad alimentaria y en el ámbito internacional agencias como el Codex Alimentarius han establecido las concentraciones o límites máximos de residuos (LMR) que debe contener un alimento para ser considerado inocuo para el consumo humano [7]-[10]. Los exportadores de productos acuícolas deben cumplir con los requisitos sanitarios de LMR, establecidos por las autoridades de los países de destino, para evitar posibles detenciones y rechazos de sus envíos debido a la presencia de residuos o contaminantes en cantidades superiores a las permitidas. Los LMR de los medicamentos veterinarios incluidos en el presente estudio varían desde 100 hasta 2000 μg/kg; asimismo, el cloranfenicol y los beta-agonistas se encuentran prohibidos para uso en la producción animal, según lo establecido por el Código de Regulación de los Estados Unidos [7], por la Regulación Europea [8], [9] y por el Codex Alimentarius [10].
Los laboratorios que realizan ensayos de residuos de medicamentos veterinarios juegan un rol importante en el control de la inocuidad alimentaria de productos de acuicultura en el mercado nacional e internacional. En el Perú, la entidad encargada de realizar los controles oficiales de residuos de medicamentos veterinarios en los productos acuícolas es el Organismo Nacional de Sanidad Pesquera, que realiza muestreos en centros de producción y en lotes de exportación, y envía las muestras a laboratorios extranjeros para su análisis, debido a la no disponibilidad de laboratorios nacionales que realicen estos ensayos.
La cromatografía líquida acoplada a espectrometría de masas en tándem (UPLC-MS/MS) es la técnica de referencia recomendada por la Unión Europea [11], [12] y por la guía de validación de la administración de alimentos y medicamentos de los Estados Unidos [13] para realizar los análisis de residuos de medicamentos veterinarios en los alimentos, debido a su alta sensibilidad y selectividad, que permiten realizar el estudio simultáneo de diferentes grupos de fármacos y/o sus metabolitos en niveles de trazas, en el orden de partes por billón y en concentraciones menores de los LMR. La UPLC-MS/MS genera información de la estructura molecular de los analitos, a partir del monitoreo de los iones productos originados por la disociación de la molécula precursora, que se utiliza para confirmar la identidad de los analitos [11]-[13]. Esta característica de la UPLC-MS/ MS ayuda a evitar o disminuir la ocurrencia de resultados falsos positivos o falsos negativos, que pueden originar problemas de detenciones o rechazos de los envíos, así como pérdidas económicas y disputas legales en el comercio internacional de alimentos. Además, la UPLC-MS/MS permite realizar análisis multirresiduo para la detección y cuantificación simultánea de cientos de moléculas en una misma corrida, sin necesidad de una rigurosa separación cromatogràfica, lo cual simplifica los procesos analíticos y facilita una rápida respuesta de los laboratorios de ensayo en el control de la inocuidad alimentaria [11].
La interferencia de matriz es una de las principales desventajas de la técnica UPLC-MS/MS, que se origina por la presencia de ciertos compuestos orgánicos de los alimentos que permanecen como impurezas en los extractos de las muestras y afectan la ionización de los analitos. Para superar esta interferencia se requiere realizar una exhaustiva limpieza de los extractos, mediante uno o más métodos de purificación, tales como la extracción en fase sólida, extracción líquido-líquido, extracción asistida por microonda o extracción acelerada por solventes. Sin embargo, los laboratorios que realizan análisis rutinarios requieren utilizar métodos sencillos con la finalidad de generar resultados rápidos para el control de la inocuidad alimentaria. La dSPE, introducida para el análisis de residuos de plaguicidas en frutos y vegetales [14], es actualmente la técnica de extracción y limpieza de muestras de mayor aceptación internacional en los laboratorios que realizan análisis para el control de la inocuidad alimentaria.
El presente estudio se realizó con el objetivo de desarrollar y validar un método de análisis sencillo, robusto y efectivo para la determinación multirresiduo de 30 medicamentos veterinarios pertenecientes a diferentes familias de fármacos (penicilinas, tetraciclinas, macrolidos, entre otros), en trucha (Oncorhynchus mykiss) y langostino (Litopenaeus vannamei). El método presentado en este artículo utiliza dSPE con C18 y un posterior análisis instrumental por UPLC-MS/MS.
Materiales y métodos
Estándares y reactivos
Para evaluar los medicamentos veterinarios de interés se utilizaron estándares de alta pureza (mayor de 99%). Los estándares de tetraciclina, clortetraciclina, oxitetraciclina, ácido oxolínico,flumequina,ácidonalidíxico, enrofloxacino, ciprofloxacino, sulfamerazina, sulfacloropiridazina, sulfadiazina, sulfametoxazol, sulfaquinolaxina, sulfametazina, sulfatiazol, sulfadimetoxina, sulfametoxipiridazina, sulfadoxina, espiramicina, eritromicina, trimetoprim, cimaterol, salbutamol, Terbutalina, ractopamina, zilpaterol, clembuterol, florfenicon y cloranfenicol fueron adquiridos de Sigma Aldrich (San Luis, EE. UU.). Los estándares de amoxicilina y mapenterol fueron adquiridos de Dr. Ehrenstorfer (Augsburg, Alemania).
Los reactivos utilizados fueron de grado LC-MS o HPLC. Metanol y acetonitrilo fueron obtenidos de Sigma-Aldrich, ácido fórmico de Fluka, sulfato de magnesio anhidro y acetato de sodio anhidro de JT Baker (Deventer, Holland) y agua ultrapura del equipo Milipore Mili-Q system (Milford, MA, EE. UU.).
Las soluciones individuales concentradas de 2000 ng/μL (soluciones stock) de cada estándar de medicamento fueron preparadas disolviendo una cantidad apropiada de cada estándar primario en acetonitrilo, metanol o agua ultrapura. Las soluciones fueron almacenadas a -20 °C en la oscuridad. Las soluciones de mezclas estándares de 40 ng/μL fueron preparadas tomando una alícuota de cada una de las soluciones stock y disolviéndolas en acetonitrilo, con excepción de los betalactámicos, en los cuales se utilizó agua para la disolución y almacenamiento en viales plásticos, a fin de evitar problemas de absorción en vidrio de los antibióticos. Las soluciones de calibración de 10 ng/μL y 2 ng/μL fueron preparadas disolviendo en acetonitrilo una cantidad apropiada de las soluciones de mezclas de estándares de 40 ng/μL.
Preparación de las muestras
Las muestras de truchas y langostinos fueron obtenidas de empresas locales dedicadas a la acuicultura. Fueron mantenidas en almacenamiento a -20 °C y protegidas de la luz hasta su preparación para el análisis. Así mismo, fueron descongeladas y llevadas a temperatura ambiente antes de realizar los ensayos. Se procedió a la molienda de langostinos (sin cabeza y antenas) y trucha (solo músculo) utilizando el homogenizador Robot coupe Blixer 3.0. Luego se realizó la extracción de la siguiente manera: se pesaron aproximadamente 2,00 g de músculo de trucha o langostino dentro de tubos de centrifuga de polipropileno de 50 mL, luego se agregaron 10 mL de acetonitrilo/EDTA 0.1 M en agua (4/1%v/v) y se homogeneizó con un equipo vortex por 5 min, luego se procedió a centrifugar por 5 min a 4000 rpm, el sobrenadante se trasvasó a un tubo de 50 mL que contenía 300 mg de adsorbente C18; luego se adicionaron 10 mL de hexano presaturado en acetonitrilo, se agitó por 30 s y se procedió a centrifugar nuevamente por 5 min a 4000 rpm. Se eliminó la capa de hexano con una pipeta Pasteur, del sobrenadante que quedó se tomó una alícuota de 5 mL y se llevó a un tubo de centrifuga de polipropileno de 15 mL. Finalmente, se concentró el extracto a sequedad, en un equipo de evaporación de nitrógeno a 45 °C. El extracto evaporado fue reconstituido con 1 mL de fase móvil A (ácido fórmico al 0,1% en agua), se filtró a través de filtros de difluoruro de polivinilo (PDVF) de 0,2 um y se colocó en viales de color ámbar. Finalmente, se inyectaron 2 μL en el equipo de UPLC-MS/MS.
Análisis por UPLC-MS/MS
El análisis por cromatografía se realizó con el equipo Acquity UPLC system (Waters, Milford, MA, EE. UU.) y la separación de los analitos se realizó con la columna Acquity UPLC BEH C18 (100 mm x 2,1 mm, 1,7 μm). Para la corrida cromatográfica se utilizó como fase móvil ácido fórmico al 0,1% en agua ultrapura (Fase A) y 0,1% ácido fórmico en acetonitrilo (Fase B) con un flujo de 0,5 mL/min. El gradiente de elución inició con 0% fase móvil B por 0,1 min, que se incrementó en forma lineal hasta 100% en 7,9 min, y se mantuvo por 1,5 min antes de regresar a la condición inicial en 0,1 min, con un tiempo de preequilibración de 3,4 min. El tiempo de la corrida cromatográfica fue de 13 min, el volumen de inyección fue de 2 μL y la temperatura de la columna fue de 40 °C.
El análisis por espectrometría de masas se realizó con el equipo UPLC-MS/MS Acquity Xevo TQ-XS (Waters, Manchester, Reino Unido). El equipo fue operado utilizando la fuente de ionización por electrospray (ESI) en modo ESI positivo. Los parámetros para la fuente de ionización fueron: voltaje de capilar 3,0 kV, temperatura de la fuente 150 °C, temperatura de desolvatación 500 °C, flujo del gas de cono 150 L/h y flujo del gas de desolvatación 1000 L/h. La colisión inducida por disociación se realizó utilizando argón como gas de colisión, a una presión 4 x mbar en la celda de colisión. La adquisición y procesamiento de datos en el equipo UPLC-MS/MS se realizó con el programa MassLynx 4.2. En la Tabla 1 se muestran los principales parámetros de espectrometría de masa MS/MS por cada analito determinado.

Validación del método
La validación del método se realizó con la finalidad de verificar el funcionamiento analítico del método desarrollado para el uso propuesto, es decir, el análisis de residuos de medicamentos veterinarios en productos acuícolas (truchas y langostinos). La validación se realizó de acuerdo al procedimiento de la Regulación Unión Europea 2002/657 [11] y Eurachem [15]. Para ello, se utilizó muestra de blancos de matriz de truchas y langostinos fortificados con cantidades apropiadas de la solución de estándares de trabajo para obtener tres niveles de concentraciones de 10, 50 y 100 μg/kg, con 10 análisis repetidos por cada nivel. Para determinar la reproducibilidad intralaboratorio, se realizaron los análisis en tres ocasiones diferentes, con tres analistas. La linealidad fue evaluada utilizando curva de calibración fortificada en blanco de matriz en 5 niveles de concentración de 10 μg/kg hasta 300 μg/kg. Los límites de deteción (LoD) y de cuantificación (LoQ) fueron estimados utilizando blancos de matriz fortificados a la concentración de 10 μg/kg, con 10 análisis repetidos.
La evaluación de la selectividad del método por UPLC-MS/MS se realizó mediante adquisición por espectrometría de masa en modo de monitoreo de reacción múltiple (MRM), con dos iones productos por cada compuesto. La proporción de las señales de los iones productos (relaciones iónicas) de cada compuesto se halló dividiendo las señales del ion menos abundante entre el ion más abundante. La identificación de los compuestos por UPLC-MS/MS se realizó según los requisitos de relación iónica establecidos por Codex [13].
La estimación de la incertidumbre se realizó de acuerdo al procedimiento establecido en la Guía ISO o método GUM [16] y Eurachem [17]. La estimación de la incertidumbre combinada se calculó a partir de la Ec. (1):

Donde C=contenido en la muestra en μg/kg, Co=concentración en el extracto en ng/mL obtenida por interpolación en la curva de calibración, Ve=volumen del extracto, W=peso de la muestra, F=factor de concentración y R=repetibilidad. La incertidumbre expandida se calculó multiplicando la incertidumbre combinada por K=2, considerando un nivel de confianza de 95%.
La evaluación de los parámetros de validación se realizó de acuerdo a los criterios de aceptación establecidos por el Codex Alimentarius [13], con RSD menor de 20% para la repetibilidad y reproducibilidad intralaboratorio y con recuperación de 70% a 120% para la veracidad. La selectividad del método se evaluó comparando los cromatogramas de los blancos de matriz versus los blancos de matriz fortificados, verificando que no hubiera señales interferentes en la región en donde eluyen los analitos. Asimismo, se realizó la confirmación por espectrometría de masas comparando la relación iónica de dos fragmentos MS/MS de cada molécula, verificando que las relaciones iónicas de las muestras estuvieran dentro del intervalo de aceptabilidad, en comparación con los estándares establecidos por el Codex Alimentarius [13].
Resultados y discusión
Análisis por UPLC-MS/MS
La optimización de los parámetros de espectrometría de masa MS/MS fue realizada mediante infusión directa en el espectrómetro de soluciones de cada uno de los estándares de medicamentos veterinarios, a la concentración de 0,5 mg/L. El electrospray fue utilizado en modo ion positivo, puesto que todos los analitos presentaban mayor señal en este modo de ionización. En la Tabla 1 se muestran las condiciones del espectrómetro de masas utilizadas en el estudio. En cuanto a la condiciones cromatográficas, fueron desarrolladas principalmente para optimizar la forma de pico, resolución e intensidad de los analitos. La fase móvil fue investigada principalmente para maximizar la sensibilidad y la resolución del método. Se probaron diferentes solventes polares, tales como metanol o acetonitrilo y agua ultrapura, todos con concentraciones de ácido fórmico al 0,05 y 0,1% (v/v). El acetonitrilo presentó mayor sensibilidad para los analitos y el ácido fórmico favoreció la ionización de los analitos. De igual manera, se configuró el tiempo de monitoreo de cada transición (dwell time) utilizando el programa MassLynx 4.2, con la finalidad de obtener como mínimo 15 puntos en cada pico cromatográfico.
En la Figura 1 se muestra el cromatograma de ion total (TIC) para la matriz trucha. Los tiempos de retención de los compuestos en las matrices de trucha y langostino fueron similares; variaron desde 1,66 hasta 4,19 minutos. La gradiente utilizada sirvió para la separación de los analitos en menos de 5 minutos. A pesar de que algunos compuestos coeluyen, ello no constituyó problema para su cuantificación, debido a la alta selectividad de la técnica de UPLC-MS/MS, puesto que estos compuestos son separados y detectados mediante adquisición de monitoreo de reacción múltiple por espectrometría de masas en tándem.
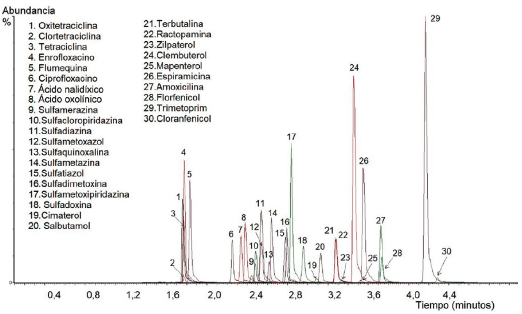
Validación del método
Después de determinar las condiciones de extracción, de separación cromatográfica y de detección por espectrometría de masas, se validó el método de análisis, determinándose los parámetros de recuperación, repetitividad, reproducibilidad intralaboratorio, linealidad, LoD, LoQ, selectividad e incertidumbre.
Recuperación
En las Tablas 2 y 3 se observa que los porcentajes de recuperación variaron desde 76,8 hasta 120,5%, valores que se encuentran dentro de la especificación establecida por el Codex Alimentarius, es decir, entre 70 y 120%.
Tabla 2 Recuperación, repetibilidad (%RSDr) y reproducibilidad intralaboratorio (%RSDR) obtenidos en trucha fortificada a 10, 50 y 100 μg/kg.

aRecuperación de 10 análisis en 03 ocasiones (n=30) (%).
bDesviación estándar relativa de 10 análisis en condiciones de repetibilidad (n = 10) (%).
cDesviación estándar relativa de 10 análisis en 03 ocasiones (n=30) (%).
Tabla 3 Recuperación, repetibilidad (%RSDr) y reproducibilidad intralaboratorio (%RSDR) obtenidos en langostino fortificado a 10, 50 y 100 μg/kg.

aRecuperación de 10 análisis en 03 ocasiones (n=30) (%).
bDesviación estándar relativa de 10 análisis en condiciones de repetibilidad (n = 10) (%).
cDesviación estándar relativa de 10 análisis en 03 ocasiones (n=30) (%).
Repetibilidad y reproducibilidad intralaboratorio
En las Tablas 2 y 3 se observa que la repetibilidad y la reproducibilidad intralaboratorio, expresadas como % RSD, variaron desde 0,6% hasta 12,8%, con lo que se cumple con los criterios del Codex Alimentarius que establecen un % RSD < 20%. Cabe destacar que dichos porcentajes se encuentran en línea con otros estudios, en los cuales se validó la técnica analítica en matrices como pescados y langostinos, en los que se obtuvieron % RSD superiores a 20 % [18]-[21].
Límites de detección y de cuantificación
El LoD y el LoQ hallados en trucha y langostinos variaron desde 0,6 μg/kg hasta 12,8 μg/kg. En la Figura 2 se observa que la mayoría de los LoQ de los medicamentos incluidos fueron menores de 10 μg/kg, a excepción de cloranfenicol y florfenicol en langostino, que fueron de 12,7 y 12,8 μg/kg, respectivamente. Dichos valores están muy por debajo de los LMR de estos compuestos, que varían desde 50 hasta 2000 μg/kg, a excepción del cloranfenicol y los compuestos beta-agonistas, medicamentos prohibidos para uso en animales para consumo humano y para los que no se han establecido sus respectivos LMR.
Los LoQ hallados para cloranfenicol fueron de 6,4 y 12,7 μg/kg para trucha y langostino, respectivamente, superiores al límite de performance mínimo requerido de 0,3 μg/kg establecido para cloranfenicol por la Regulación Europea [6], lo que se explicaría por el hecho de que, al haberse utilizado un método multirresiduo, no se puede aplicar condiciones de sintonización del espectrómetro de masas, condiciones cromatográficas y condiciones de extracción especificas con la finalidad de incrementar la sensibilidad del método para esta molécula. Por ello, lo recomendable sería realizar un método individual para el análisis del cloranfenicol, con la finalidad de obtener límites analíticos menores o iguales a lo requerido. Una situación diferente se observó para el florfenicol, para el cual se obtuvo LoQ de 7,2 y 12,8 μg/kg para trucha y langostino, respectivamente, similares al cloranfenicol, por lo que estos límites analíticos obtenidos serian adecuados para el análisis de florfenicol, debido a que están muy por debajo del LMR, que es de 1000 μg/kg. Hay que remarcar que en investigaciones previas se han reportado valores de LoD y LoQ similares o por debajo de lo hallado en el presente estudio [18]-[26].

Linealidad
Los coeficientes de determinación (R2) de las curvas de calibración de los medicamentos veterinarios fortificados en trucha y langostino fueron menores de 0,99; con excepción de sulfaquinoxalina en langostino, que fue de 0,97. En la Figura 3 se observan las curvas de calibración para la matriz trucha de compuestos representativos de los principales grupos de fármacos veterinarios incluidos en el estudio de validación.

Selectividad
La selectividad del método se evidenció mediante identificación por LC-MSMS de los medicamentos veterinarios incluidos en el estudio, dado que las relaciones iónicas de los compuestos cumplieron con los rangos de aceptación del Codex [13]. En la Figura 4 se muestran los cromatogramas MRM de los compuestos representativos fortificados en blando de matriz trucha, en los que se observa que el monitoreo de dos iones fragmentos característicos de la estructura molecular de cada compuesto le confiere una alta selectividad a la técnica de UPLC-MS/MS. Asimismo, se identificaron las moléculas por el tiempo de retención (TR). Por otro lado, se observó efecto de arrastre en los compuestos del grupo de las quinolonas, vale decir, ácido nalidíxico, ácido oxolínico, enrofloxacina y ciprofloxacina. Para superar este efecto, se requeriría investigar los métodos de lavado del inyector utilizando diferentes solventes y tiempos de lavado. Sin embargo, debe puntualizarse que el efecto de arrastre fue insignificante y no afectó a la cuantificación de dichos compuestos en las matrices de langostino y trucha.
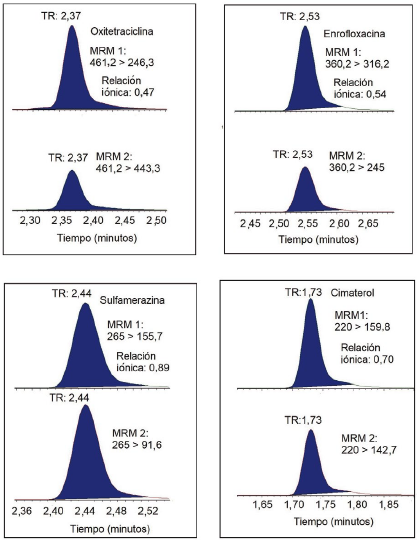
Incertidumbre
La incertidumbre expandida de los medicamentos veterinarios incluidos en el estudio de validación varió desde 8% a 59% para trucha y langostino, con excepción del compuesto sulfaquinoxalina en langostino, para el que se obtuvo un valor de 98%, debido principalmente a la alta contribución de la incertidumbre de la curva de calibración, tal como se observa en la Figura 5, lo que se originó probablemente debido a errores aleatorios o sistemáticos en la preparación de la curva de calibración.

Análisis en muestras reales
Una vez desarrollado el método, fue aplicado para la determinación de residuos de medicamentos veterinarios en 6 muestras de trucha, las cuales fueron recolectadas en 6 supermercados diferentes del Cercado de Lima, Perú. Con el fin de asegurar la calidad de los resultados, se fortificó una muestra blanco a la concentración de 50 μg/kg antes de comenzar con la extracción. Además, el tiempo de retención, los fragmentos de cuantificación y confirmación y el ratio iónico encontrados en las muestras de mercado fueron comparados con los de los estándares de calibración, para su correcta identificación y cuantificación, tal como lo establece la Decisión de la Comisión 657/2002/ CE [11]. Se reportaron trazas del medicamento oxitretraciclina en una de las 6 muestras, la cual se encontraba muy por debajo del LMR de 100 μg/kg. Dichos resultados concuerdan con otros trabajos de investigación reportados previamente utilizando la misma técnica analítica, en los que se detectaron pocos analitos en muestras de trucha [18]-[21].
Conclusiones
El estudio cumplió con el objetivo de desarrollar y validar un método de análisis sencillo, robusto y efectivo para la determinación multirresiduo de múltiples medicamentos veterinarios pertenecientes a diferentes familias de fármacos, en trucha y langostino. El método, que utiliza dSPE con C18 y detección por cromatografía líquida acoplada a espectrometría de masas, cumplió con los criterios de validación establecidos por el Codex Alimentarius. Los resultados obtenidos nos permiten recomendar la aplicación del método en los programas nacionales de monitoreo de la inocuidad de truchas y langostinos provenientes de acuicultura.

