











 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
Permalink
Introduction
Autoimmune hepatitis (AIH) is characterized as an immune-mediated inflammatory disorder lacking distinctive diagnostic markers. Its diagnosis necessitates histological anomalies, specific laboratory findings, autoantibody positivity, and the exclusion of alternative hepatic conditions such as viral hepatitis, hereditary diseases, metabolic and cholestatic disorders, among others1,2. The diagnostic challenge in AIH stems from its broad spectrum of clinical presentations, the absence of pathognomonic features, and variability in the treatment outcomes3.
Case presentation
This report details the case of a 28-year-old female patient with no personal pathological history, medication or hepatotoxin use, alcohol consumption, other toxic habits, or a family history of autoimmune diseases. She presented with a week-long progression of cutaneous jaundice, mucosal and scleral icterus, associated with diffuse and intermittent abdominal pain. Persistent jaundice and the onset of generalized itching prompted her emergency department visit. Upon admission, she exhibited normal vital signs, and the physical examination revealed generalized jaundice and abdominal pain upon deep palpation in the upper right quadrant, with the remainder of the physical examination showing no abnormalities.
Initial laboratory tests did not reveal any abnormalities beyond those described in Table 1. The R factor was calculated (Formula: patient’s ALT/upper normal limit of ALT ÷ patient’s alkaline phosphatase/upper normal limit of alkaline phosphatase) to assess the predominant alteration pattern in liver tests, yielding a result of 17.6, indicative of hepatocellular injury (R factor > 5 denotes hepatocellular injury, < 2 cholestatic injury, and 2-5 a mixed pattern)4. General urine examination (GUE) reported positive urobilinogen, while inflammatory markers such as procalcitonin and C-reactive protein (CRP) were found to be negative.
Table 1 Liver Tests Before and After Initiation of Corticosteroid Therapy
ALT: Alanine Aminotransferase; AST: Aspartate Aminotransferase; DB: Direct Bilirubin; IB: Indirect Bilirubin; TSB: Total Serum Bilirubin; ALP: Alkaline Phosphatase; GGT: γ-Glutamyl Transferase; IgG: Immunoglobulin G; INR: International Normalized Ratio; NR: Not Reported. Author’s own research.
An abdominal ultrasound was conducted, yielding the following results: the liver exhibited normal shape and size, with no discernible abnormalities in the gallbladder or bile ducts. In the color Doppler assessment, intrahepatic vessels demonstrated unremarkable perfusion. Subsequent testing for hepatotropic viruses, including anti-HAV IgM, HbsAg, ELISA Anti-HCV, anti-HBc IgM, and anti-HEV IgM, returned negative results. Similarly, a panel testing for non-hepatotropic viruses, comprising herpes virus, varicella-zoster virus, cytomegalovirus, and Epstein-Barr virus, also showed negative findings for both IgG and IgM. Additionally, a fourth-generation rapid test for HIV was non-reactive. Autoantibody screening for autoimmune hepatitis encompassed ANA, SMA, anti-LKM1, anti-LC1, and anti-SLA antibodies, all of which were negative. Quantification of IgG levels revealed a value of 4316.1 mg/dL, surpassing the reference range of 600-1600 mg/dL.
Following these investigations, a percutaneous liver biopsy was undertaken, yielding histological features consistent with AIH (Figure 1). These included a lymphoplasmacytic infiltrate in the portal and periportal regions, indicative of interface activity, as well as lobular and hepatic necrosis, pericentral necrosis, and the absence of ductal lesions. Masson’s trichrome staining failed to reveal collagen deposition. Utilizing the revised International Autoimmune Hepatitis Group (IAIHG) scoring system from 19995, a pre-treatment score of 16 points was calculated, affirming the diagnosis of definitive AIH.
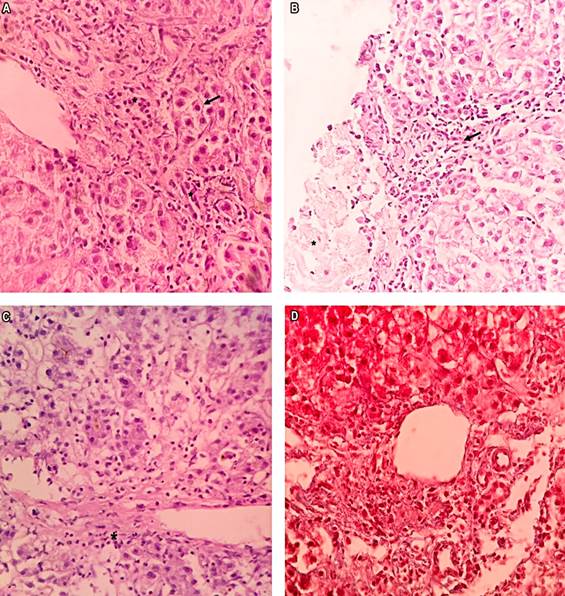
Figure 1 A. *: Interface activity. Arrows: Hepatocytes with a regenerative appearance (hematoxylin and eosin [H/E] stain: 20x magnification). B. *: Moderate necroinflammation (grade 2) at the lobule level. Arrow: Lymphoplasmacytic inflammatory infiltrate (H/E stain: 20x magnification). C. *: Inflammation and necrosis surrounding a central vein. Centrolobular interface necrosis (grade 1). D. Masson’s trichrome staining at 20x magnification. No collagen deposition, stage 0. Activity at grade I. D. Basal membranes of the biliary ducts preserved.
Treatment was commenced following the clinical guidelines of the American Association for the Study of Liver Diseases (AASLD)6. Prednisone was administered at a dosage of 60 mg/day, and the patient responded favorably, exhibiting clinical and biochemical improvement. This was evidenced by the normalization of the international normalized ratio (INR) and a decrease in transaminase levels by day three of corticosteroid therapy. After seven days of hospitalization, the patient was discharged, and azathioprine at a dose of 50 mg was introduced at week two of treatment. At the three-month follow-up, the patient remained asymptomatic and demonstrated biochemical remission, with liver tests and IgG values within normal ranges (Table 1).
Discussion
HAI manifests worldwide, transcending ethnic and age boundaries with a bimodal distribution pattern: one peak occurs between ages 10 and 18, and another surfaces between the fourth and sixth decades of life. The condition predominantly affects females, with a 3:1 female-to-male ratio. Global incidence rates are estimated to range from 0.67 to three cases per 100,000 person-years2,6,7. According to a recent Colombian retrospective cohort study, 80% of patients were female with a median age of 49 years8.
The precise pathophysiological mechanisms leading to hepatic autoimmunity are yet to be fully understood. Current hypotheses suggest a multifactorial etiology, encompassing genetic and epigenetic factors, aberrant autoregulatory immune mechanisms, and environmental precipitants7,9. The breakdown of self-tolerance to hepatic autoantigens triggers an immune reaction. Regulatory T cells (Tregs) fail to suppress the autoimmune response to these antigens when confronted with environmental factors, such as viral infections or xenobiotics. Antigen-presenting cells (APCs) then present autoantigenic peptides to naive Th CD4+ cells through α/β T cell receptors (TCRs). Specific cytokine secretions prompt B cells to produce autoantibodies, lead to the development of regulatory B cells (Bregs), activate macrophages, and instigate pathogenic cytotoxicity via Th17 CD4+ cells. The resulting infiltration of inflammatory effector cells leads to periportal and lobular cytotoxicity, necroinflammatory hepatocyte destruction, and the activation of periportal stellate cells. These cells amplify the local immune response and drive the progression of portal fibrosis, which, without immunosuppressive treatment, can progress to cirrhosis6,7.
The absence of defining characteristics complicates the diagnosis of HAI, relegating it to one of exclusion. The clinical manifestation spectrum is broad and varies by race, from asymptomatic liver disease to severe hepatitis presentations6,7. Acute onset (< 30 days) occurs in about 25%-75% of patients, with 3%-6% developing acute liver failure2. The majority will have chronic, nonspecific symptoms like fatigue, malaise, and arthralgia before presentation. The physical exam may be unremarkable or occasionally reveal hepatomegaly, painful splenomegaly, and, in advanced cirrhosis, clinical signs of chronic liver pathology, such as palmar erythema, spider angiomas, and gynecomastia2,6,7.
Autoantibodies serve a diagnostic purpose during acute HAI episodes3. ANAs are present in 80%, SMAs in 63% and anti-LKM1 in 3% of cases6. Isolated serological findings of ANA, SMA, or anti-LKM1 occur in 49% of patients, while 51% possess multiple antibodies. ANAs are either undetectable or present in low titers in 30%-40% of patients10. Serum IgG levels are found to be normal in 25%-39% of the cases3,6,7.
Circulating autoantibodies are absent in approximately 10% of patients with autoimmune hepatitis11-13. The prevalence of seronegativity is subject to regional variation and study design: for instance, in a cohort of 167 Chinese patients, 10.2% were seronegative10; in a study of 278 patients in Medellín, the seronegativity rate was 8.6%14; whereas a European study on 126 patients with cryptogenic hepatitis found that 34% were classified as seronegative AIH based on the IAIHG diagnostic score15. Seronegativity may arise from the inability of current assays to detect antigen-antibody complexes, the presence of antibodies not yet identifiable by commercially available tests, or fluctuations in antibody levels13. Seronegative AIH patients tend to present more frequently with severe disease compared to their seropositive counterparts (50% vs 20.3%), possibly due to immune dysfunction secondary to liver failure; however, the response to treatment generally parallels that of seropositive AIH16.
In AIH, liver biopsy remains an essential and obligatory procedure for confirming diagnosis, assessing the severity and the level of inflammatory activity, and ruling out other liver diseases. Interface hepatitis stands out as a distinguishing feature in AIH. According to the 2008 simplified IAIHG criteria, the presence of interface hepatitis alone aligns with an AIH diagnosis, and this is often accompanied by a plasma cell infiltration in 66% of cases, emperipolesis in 65%, and lobular hepatitis in 47%6,17,18.
To aid and standardize AIH diagnosis, various diagnostic scoring systems have been proposed. The original IAIHG scale, introduced in 199319 and revised in 19995, was supplemented by a simplified scoring system published in 200820. The 1993 scale was deemed too intricate, incorporating clinical, biochemical, histological, and genetic parameters, and evaluating the response to treatment19. The 1999 revision, although still complex, struggled to effectively distinguish between AIH and cholestatic syndromes21.
The 2008 simplified score offers a pragmatic, clinic-friendly alternative that relies on just four parameters: autoantibodies, hypergammaglobulinemia, histology, and exclusion of viral hepatitis20. The simplified and revised scores have comparable diagnostic accuracies in patients exhibiting classical AIH features; nonetheless, the simplified score may not suit cases with atypical features or seronegativity, for which the more comprehensive 1999 revised score is advised 22,23. The latter is also suggested to be more effective for diagnosing acute onset AIH with severe presentation3.
Conclusions
AIH is a relatively rare clinical entity posing a significant diagnostic challenge. It necessitates the assimilation of congruent clinical, paraclinical, and histopathological evidence. Diagnostic scales like the IAIHG’s revised or simplified versions contribute to the standardization of the diagnostic process. Effective management with corticosteroids and immunosuppressive agents facilitates remission of hepatic inflammation and hepatic fibrosis regression, thereby averting progression to advanced cirrhosis and its attendant complications.

