











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
PermalinkEl síndrome respiratorio agudo severo causado por el coronavirus tipo 2 (SARS-C0V-2), descubierto en Wuhan (China) en el 2019, culpable de la actual pandemia de COVID-19, ha causado millones de casos confirmados en humanos de todo el mundo y otros cuantos confirmados en animales tanto domésticos como silvestres 1-5. También ha generado un sin número de problemáticas sociales, económicas, culturales, etc., en todo el mundo. Por ello, es importante abordar esta problemática desde otras disciplinas que complementen el enfoque epidemiológico predominante 6.
Desde el inicio de la pandemia en el 2020 se sospechó de su origen zoonótico, tal como se confirmó posteriormente, pese a que aún no se tiene total claridad del papel que pueden jugar los animales silvestres y domésticos en la cadena de transmisión entre las especies animales y los seres humanos y viceversa 7,8.
Los coronavirus han sido protagonistas de otras pandemias años atrás y han representado una preocupación constante en el mundo de la salud animal por más de treinta años, durante los cuales se han llevado a cabo múltiples investigaciones para entender el comportamiento de estos virus y cómo sería el mejor manejo, tratamiento o vacuna (si es el caso). Pese a todos estos esfuerzos, muchos coronavirus no se han podido eliminar, aunque y sí se han podido controlar parcialmente 9-11, como el caso del coronavirus felino (FCoV) causante de la peritonitis infecciosa felina (PIF), una patología que no solo afecta a los felinos domésticos, sino al parecer a felinos y otros animales silvestres. Sin embargo, aún existen muchos vacíos del conocimiento respecto a esta enfermedad que se sigue investigando alrededor del mundo y los resultados que se han obtenido a través del tiempo podrían ayudar al entendimiento de los casos de SARS-CoV-2 actuales desde lo clínico 12-16.
A la fecha se han publicado más de 1000 artículos científicos sobre el SARS-CoV-2 en humanos. No obstante, aún existen varios interrogantes sobre este virus 1-16. En contraste, es mucho menor el número de publicaciones sobre el SARS-CoV-2 en animales domésticos y silvestres, lo cual constituye un reto para la salud pública.
En el caso específico de la identificación molecular de SARS-CoV-2 en animales hay muy pocas publicaciones, pese a que el virus se ha encontrado en tigres y leones (siete animales en total del Zoológico del Bronx en Nueva York), en primates no humanos, en trece gatos y en cuatro perros alrededor del mundo. Además, en estudios experimentales han resultado susceptibles los felinos, los hurones, primates y cerdos principalmente 3-11,13-15.
MÉTODOS
Descripción del caso
La identificación molecular de SARS-CoV-2 se realizó en una leona africana (Panthera leo), de 16 años, que vivía bajo cuidado humano en un zoológico colombiano. Los signos que podrían estar relacionados con el cuadro se reportaron en 2021 (Figuras 1 y 2).
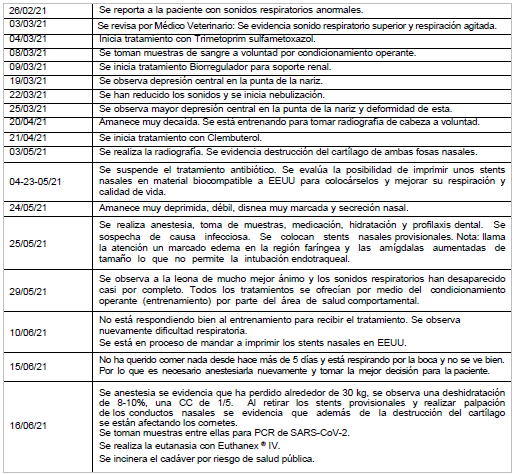
Fuente: historia clínica de la leona.
Figura 1 Resumen descripción del caso de la leona africana (Panthera leo), 2021
Revisión de información
Se realizaron diferentes búsquedas en inglés y español en buscadores como SCOPUS, Medline, Scielo, BVS, PubMed y Google Académico. Los descriptores usados fueron "coronavirus", "felinos", "SARS-CoV-2", "animales domésticos", "animales silvestres", "felinos silvestres", "humanos", "identificación molecular", "COVID-19".
Muestras clínicas
Las muestras de sangre de la leona fueron tomadas con los protocolos descritos por la medicina veterinaria para toma de muestras y se enviaron refrigeradas a un laboratorio veterinario avalado para su procesamiento.
Toma y transporte de la muestra de hisopado nasal
Las muestras de hisopado nasal se tomaron con un hisopo estéril haciendo movimiento rotatorio en cada fosa nasal, introduciendo lo más posible el hisopo. Cada hisopo fue depositado en cada medio de transporte haciendo movimientos rotatorios y dejando la punta del hisopo sumergido en cada medio de trasporte viral, que se preparó en la sala de medios de cultivo del Departamento de Microbiología de la Facultad de Medicina de la Universidad Nacional de Colombia, de acuerdo con el protocolo suministrado por el Instituto Nacional de Salud. Brevemente, para 400 ml de Caldo BHI, se agregaron 27 ml de albúmina bovina fracción V (7,5%), 4 ml de penicilina 10,000 unidades/ml y 3,2 ml de anfotericina B 250 ug/ml. Una vez depositados los hisopos en los medios de transporte correspondientes y se llevó bajo refrigeración de inmediato al laboratorio del Departamento de Microbiología de la Facultad de Medicina de la Universidad Nacional de Colombia.
Extracción de ARN viral del SARS-Cov2
El kit empleado para la extracción del ARN viral fue MGIEasy Nucleic Acid Extraction Kit39, el cual hace uso de la tecnología de perlas supermagnéticas que se unen a los ácidos nucleicos. Inicialmente se empleó un tampón de lisis para romper las células y la cápsula viral, seguido de una separación magnética del ADN/ARN. Luego se hicieron tres lavados antes de eluir el ADN/ARN en agua libre de RNAasas. A continuación, se llevó a cabo el protocolo resumido, seguido de las instrucciones del fabricante: se agregaron 200 pL de muestra a la mezcla de tampón de lisis preparada previamente (perlas supermagéticas, proteinasa K, etanol y solución MLB, como lo indica el fabricante), y se realizó vórtex durante 1 min. Seguidamente se centrifugó a 11000 rpm por 1 min e inmediatamente se colocó en el soporte magnético durante 1 min. Después de que el líquido aclaró, se desechó el líquido sobrenadante. Se añadieron 500 μl de tampón MW1 y se retiró el tubo del soporte magnético; luego se mezcló bien durante 5-10 s. El tubo de centrífuga se colocó de nuevo en el soporte magnético durante 1 min. Una vez que el líquido quedó completamente claro, se desechó el sobrenadante. Luego se añadieron 500 μL de tampón MW2. Se retiró el tubo del soporte magnético y se mezcló bien durante 5-10 s. Se colocó el tubo de centrífuga en el soporte magnético durante 1 min y se desechó el sobrenadante como se indicó anteriormente. El siguiente paso fue agregar 600 pL de etanol absoluto y se retiró el tubo de centrífuga del soporte magnético. Se mezcló bien durante 5-10 s y se colocó de nuevo en el soporte magnético durante 1 min. Una vez que el líquido quedó completamente claro, se desechó el sobrenadante. Luego se incubó a 56°C hasta que se evaporó todo el etanol. Luego se retiró el tubo de centrífuga del soporte magnético y se adicionaron 50 μL de agua libre de RNasa. Se mezcló y se incubó a 56°C, 1200 rpm durante 3 min. Después de los 3 min, se colocó el tubo de centrífuga en el soporte magnético. Después de que el líquido quedó completamente transparente, se transfirió con cuidado 47 μL de la solución de ARN a un tubo de 1,5 ml. Se etiquetó y se almacenó a -25 °C.
Detección de los genes E y RdRp del virus SARS-Cov2 mediante tiempo real RT-qPCR
Se empleó la tecnología TaqMan estandarizada por el grupo de virología de Charité-Berlin 40 con algunas modificaciones. Los iniciadores y sonda utilizados para la detección de los genes E y RdRp fueron los diseñados por el mismo grupo de virología de Charité-Berlin para la detección del ARN viral 17. Como control de la extracción para detectar falsos negativos debido a inhibidores de la muestra, se empleó el gen humano de la ribonucleasa P (RNasa P), cuya detección fue estandarizada por el Grupo de Biología Molecular de la Secretaría de Salud de Bogotá (comunicación personal). Los iniciadores y sondas de los genes analizados se muestran en la Tabla 1.
Tabla 1 Iniciadores y sondas empleadas en la detección de SARS-Cov1
| Nombre del primer sonda | Secuencia de los primers/Sondas 5'-3* |
|---|---|
| RNAse P For. | AGATTTGGACCTGCGAGCG |
| RNAse P Rev. | GAGCGGCTGTCTCCACAAGT |
| RNAse P Pro1 | FAM-TTCTGACCT-Nova-GAAGGCTCTGCGCG-BHQ-1 |
| RdRp_SARSr-F2 | GTGARATGGTCATGTGTGGCGG |
| RdRp_SARSr-R1 | CARATGTTAAASACACTATTAGCATA |
| RdRp SARSr-P2 | FAM-CAGGTGGAACCTCATCAGCGAGATGC-BHQ-1 |
| E-Sarbeco-F1 | ACAGGTACGTTAATAGTTAATAGCGT |
| E-Sarbeco-R2 | ATATTGCAGCAGTACGCACACA |
| E-Sarbeco-P1 | FAM-ACACTAGCCATCCTTACTGCGCTTCG-BHQ-1 |
Se empleó la mezcla Luna Universal Probe One Step (New England Biolabs) para la amplificación. La concentración de reacción de los iniciadores para los genes E y RdRp es de 0,4 μM y de la sonda de 0,2 μM. Para el gen RNAasaP, la concentración de los iniciadores es de 0,2 μM y la sonda de 0,15 μM. El volumen de la reacción de amplificación fue de 20 μL, de los cuales 5 μL corresponden al ARN extraído. Posteriormente se colocó en el termociclador y el programa de ciclaje fue el siguiente: 50°C 15' (obtención del cDNA), seguido de 40 ciclos de 95°C5', 60°C45"; la captura de la fluorescencia se realiza en el paso de extensión, es decir, durante los 6o°C 45''. Las sondas E y RdRp son detectadas en el canal FAM y la sonda de RNasa P es detectada en el canal HEX. Las muestras fueron consideradas positivas cuando la fluorescencia sobrepasa el umbral de detección en un Ct menor a 38. De lo contrario, fueron consideradas negativas.
Secuenciación del genoma
La biblioteca de secuenciación de Nanorope se preparó de acuerdo con el protocolo de secuenciación ARTIC nCoV-2019 v3 (LoCost) (https://www.protocols.io/view/ncov-2019-sequencing-protocol-v3-locost-bh42j8ye). La muestra se transcribió inversamente con el kit LunaS-cript® RT SuperMix (New England BioLabs), y los 2 grupos de amplicones de virus de PCR se prepararon con paneles de cebadores IDT ARTIC nCoV-2019 V3. Se utilizaron el kit EXP-NBDi96-Oxford Nanopore y el kit SQK-LSK109-XL-Oxford Nanopore para el código de barras y la ligadura, respectivamente. Se usaron perlas magnéticas Ampure XP (Angencourt Bioscience Corporation, MA, EE. UU.) en los pasos de limpieza, y se usó el kit de ADN Qubit HS (Thermo Fisher Scientific Inc.) para las cuantificaciones de grupos y bibliotecas. La biblioteca se cargó y secuenció en un secuenciador MinlON MkiC (Oxford Nanopore), usando una celda de flujo FLO-MIN-106D. MinKNOW, versión 21.05.8 realizó la amplificación de bases rápidas y demultiplexación en vivo.
Análisis bioinformático
Las lecturas obtenidas fueron preprocesadas, ensambladas y analizadas por medio de la canalización Viralrecon 18. Brevemente, la calidad de la secuencia se determinó utilizando PycoQC. Luego se procesó posteriormente con el software Guppyplex y Artic Minion. Las métricas de alineación se obtuvieron utilizando SAMtools y Mosdepth. El análisis posterior para la determinación de variantes y la clasificación taxonómica se realizó con BCFtools, snpEff, Pangolin 3.1.16 y Nextclade 1.7.0. La implementación del algoritmo BLAST de NCBI y el software MAUVE también se utilizaron para análisis comparativos del genoma 19.
RESULTADOS
Revisión de información
Con los descriptores mencionados en el apartado de metodología, se obtuvieron más de 2 000 artículos discriminados de la siguiente manera:
Para descriptores como SARS-CoV-2, COVID-19 en humanos: más de 1 000
Para descriptores como SARS-CoV-2 en felinos y animales silvestres: 90
Para descriptores como coronavirus en felinos: más de 1000
Para descriptores como SARS-CoV-2 y zoonosis: 300
Luego de la lectura de los artículos, se escogieron -de acuerdo con su pertinencia y calidad- solo 140 para el desarrollo de este artículo.
Muestras clínicas
Los resultados obtenidos de las muestras de sangre se muestran de manera comparativa en la Tabla 2.
Tabla 2 Resultados comparativos de las muestras de sangre en una leona africana (Panthera leo), 2021
| Parámetro | Resultado F1* | Resultado F2** | Resultado F3*** | Unidad | Valor normal **** |
|---|---|---|---|---|---|
| Hematocríto | 44 | 38 | 42,8 | % | (24-51,5) |
| Hemoglobina | 16,9 | 13,2 | 12,5 | gr/dl | (11-17,7) |
| Eritrocitos | 12.3 | 8.7 | 8,42 | x106 cel/μl | (6,2-15) |
| VCM | 46 | 43,7 | 50,8 | fl | (37-58) |
| HCM | 13,8 | 11 | 14,8 | pg | (15-17) |
| Leucocitos | 9,4 | 14,9 | 21,1 | x103 cel/μl | (5-19,9) |
| Neutrófilos Relativo | 80 | 65 | 56 | % | (47-87) |
| Neutrófilos Absoluto | 7,5 | 9,6 | 11,8 | x103 cel/μl | (0,01-15,2) |
| Linfocitos Relativo | 20 | 15 | 25 | % | (8-36) |
| Linfocitos Absoluto | 2 | 2,2 | 5,2 | x103 cel/μl | (0-3,85) |
| Eosinófilos Relativo | 0 | 0 | 5 | % | (0-9) |
| Eosinófilos Absoluto | 0 | 0 | 1,05 | x103 cel/μl | (0,06-1,2) |
| Monocitos Relativo | 0 | 14 | 14 | % | (1-11,6) |
| Monocitos Absoluto | 0 | 2,0 | 2,95 | x103 cel/μl | (0-1) |
| Plaquetas | 536 | 464 | 488 | x103 cel/μl | (150-500) |
| Proteínas totales | 8 | 6,6 | 7,8 | gr/dl | (6,8-8,7) |
| Albúmina | - | 3,2 | - | gr/dl | (3-4,4) |
| Globulina | - | 3,4 | - | gr/dl | (3-4,4) |
| Creatinina | 3,6 | 2,3 | 3,1 | mg/dl | (1,8-3,6) |
| Nitrógeno ureico | 35 | 23 | 21,2 | mg/dl | (19,4-51,4) |
| ALT | 36 | 51 | 23 | U/L | (25-98) |
| AST | 34 | 54 | 39 | U/L | (17-59) |
| FeCoV ELISA | - | Negativo | - | - | - |
| Observaciones al | No | No | Neutrófilos tóxicos (30%) y | ||
| Cuadro Hemático | abundantes agregados plaquetarios |
Fuente: Historia clínica, Zims: 2021. F1: 08/03/21 * Bajo condicionamiento operante a voluntad; F2: 25/05/21 ** Primera anestesia; F3: 16/06/21 *** Segunda anestesia. **** Valores de referencia tomados de Zims medical para Panthera leo: 2021.
Extracción de ARN viral del SARS-Cov2
Se extrajo ARN viral compatible con SARS-Cov2 de las muestras de hisopada nasal de la leona (Panthera leo).
Detección de los genes E y RdRp del virus SARS-Cov2 y del gen RNasa P mediante tiempo real RT-qPCR
Se logró amplificar el gen E y el gen RdRp del virus SARS-Cov2 de la muestra obtenida de la leona, como se observa en las Figuras 3A y 3B, respectivamente. Igualmente, se logró detectar el gen RNasaP en el control positivo, indicando la ausencia de inhibidores de la PCR (Figura 3C).
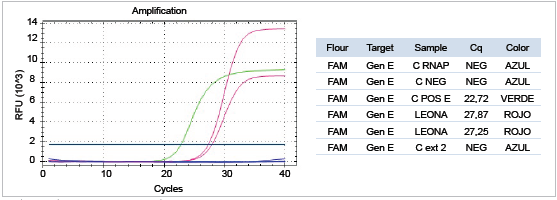
Fuente: Laboratorio de Microbiología. Facultad de Medicina. Universidad Nacional de Colombia, 2021.
Figura 3A Detección del gen E del virus SARS-Cov2 mediante tiempo real RT-qPCR de la muestra de hisopado nasal de la leona (Panthera leo), 2021
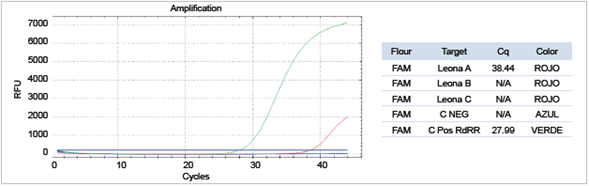
Fuente: Laboratorio de Microbiología. Facultad de Medicina. Universidad Nacional de Colombia; 2021.
Figura 3B Detección del gen RdRp del virus SARS-Cov-2 mediante tiempo real RT-qPCR de la muestra de hisopado nasal de la leona (Panthera leo), 2021
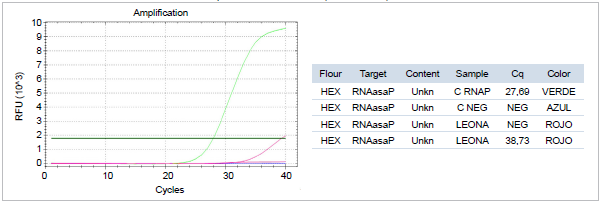
Fuente: Laboratorio de Microbiología, Facultad de Medicina, Universidad Nacional de Colombia, 2021
Figura 3C Detección del gen humano RNasa P mediante tiempo real RT-qPCR de la muestra de hisopado nasal de la leona (Panthera leo), 2021
Como control positivo para el gen E se empleó una muestra humana positiva para SARS-Cov2 y cuya curva se observa en la gráfica de color verde. El control negativo es la mezcla de PCR sin adición de cDNA (curva de amplificación de color azul). El control de extracción (C ext 2) es una muestra humana negativa para SARS-Cov2 (curva de amplificación azul). La muestra de la leona se extrajo por duplicado y se obtuvieron, en ambos casos, un resultado positivo representado por las curvas de amplificación de color rojo.
Como control positivo para el gen RdRp, se empleó una muestra humana positiva para SARS-Cov2, y cuya curva se observa en la gráfica de color verde. El control negativo es la mezcla de PCR sin adición de cDNA (curva de amplificación de color azul). La muestra de la leona se corrió por triplicado y en una de las corridas se obtuvo una curva de amplificación positiva en el límite de detección (curva de color rojo).
El control positivo para el gen RNasaP es una muestra humana, cuyo resultado fue positivo confirmado por la presencia de la curva de amplificación de color verde. Este resultado indica que no se encontraron inhibidores de la PCR en esta muestra control. En la muestra de la leona el resultado fue negativo (curvas de amplificación de color rojo). Sin embargo, fue positiva para el gen E (Figura 4A), indicando que no hubo presencia de inhibidores de la PCR.
Análisis bioinformático
Para evaluar el origen taxonómico del ARN muestreado, se usaron dos genomas diferentes como referencia para el ensamblaje. El primero fue el aislamiento del coronavirus 2 del síndrome respiratorio agudo severo Wuhan-Hu-i (número de identificación NC_045512.2 en la base de datos refseq del NCBI). El segundo fue el virus de la peritonitis infecciosa felina (FIP) (número de identificación NCJO02306.3 en la base de datos refseq del NCBI), un coronavirus que provoca una infección mortal en los felinos 20,21. El ensamblaje del genoma contra la referencia FIP no superó los requisitos mínimos de cobertura de Viralrecon y, por lo tanto, no pudo ser utilizado para un análisis posterior.
Con respecto al ensamblaje del genoma de Wuhan-Hu-1, el contenido general de N en el genoma secuenciado fue del 64%, lo que llevó a Pangolin a rechazar cualquier clasificación. Sin embargo, Nextclade pudo clasificar la secuencia del genoma como perteneciente al clado 20C e identificó las siguientes 4 mutaciones en los genes que codifican las proteínas Spike, ORF1b y ORF3a: C14408T, C23604A, G24410A y G25563T, correspondientes a las sustituciones de aminoácidos: S:P681H, S :DO,5ON, ORF1b:P314L y ORF3a:Q57H respectivamente 20,21.
Para comprender las diferencias entre el ARN de la leona muestreado y ensamblado, el genoma de FIP y el genoma de referencia de Wuhan, se realizaron análisis adicionales entre estos tres. Los análisis BLAST se realizaron entre FIP y el genoma ensamblado de la leona, así como entre este último y el genoma de referencia de Wuhan (NCJ045512.2).
La alineación general de la leona vs. FIP mostró una cobertura total del 5%, con un 68,48% de identidad (archivo complementario X, parte A), localizada en una región específica entre las posiciones ~ 15000-210000 pb en FIP. La alineación entre la leona y NCJ045512.2 mostró una cobertura del 35% con una identidad del 100%, distribuida de principio a fin de ambas secuencias (archivo complementario X, parte A). La alineación entre FIP y NC_045512 mostró una cobertura del 25%, con una identidad del 63,34% en una región localizada entre las posiciones ~10500-230oobp en el genoma de referencia de NC_045512 (archivo complementario X, parte A).
Se realizó una alineación múltiple comparativa entre los tres genomas por medio de Mauve 21, centrándose en los genes Spike, E y M (archivo complementario, parte B). Como se esperaba del análisis BLAST anterior, la FIP general parece estar más distante (en términos de similitud de secuencia) de NCJ045512 y el genoma de la leona. Con respecto al gen de codificación de Spike, se logró identificar 5 segmentos muy similares entre la leona y NCJ045512, y un segmento similar de bajo nivel entre la región de inicio de la región S, así como otros 3 segmentos de bajo nivel en la región central de la misma región S entre los tres genomas. En cuanto al gen E, no se encontró similitud entre los tres genomas, pero sí entre los genomas de leonas y NC_45512. Una situación similar se observó para el gen M.
DISCUSIÓN
El objetivo fue identificar SARS-CoV-2 en una leona africana (Panthera leo), hembra de edad avanzada, que presentó por varios meses signos de enfermedad respiratoria atípica.
La brecha que existe entre las publicaciones de humanos y animales relacionadas con la pandemia es muy amplia 1,3-11,13-15,22-24 y esto evidencia, por un lado, que la preocupación por la supervivencia de la humanidad es de vital importancia, pero, por otro lado, sigue evidenciando la falta de articulación interinstitucional e interdisciplinaria para el entendimiento y afrontamiento de enfermedades como estas, que son catalogadas como zoonosis 25,26, y el enfoque predominante epidemiológico a nivel mundial para responder a estos problemas de salud pública 6.
Lo anterior se convierte en una limitación importante para lograr una comprensión integral de la problemática actual y, al mismo tiempo, aumenta los vacíos del conocimiento en la relación salud animal-humana, atrasando el diagnóstico oportuno en el caso de los animales domésticos y silvestres y en la formulación de planes de control más eficaces para la humanidad.
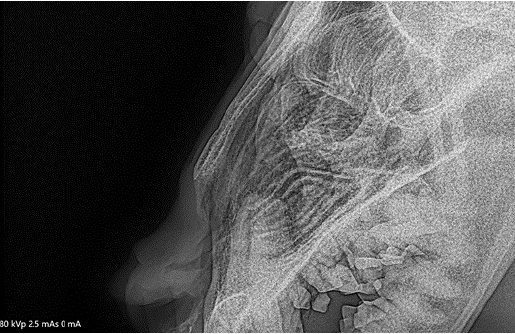
Pasando a la descripción del caso de la leona, cabe resaltar que luego de la revisión de los artículos publicados sobre SARS-CoV-2 en animales domésticos y silvestres, se puede decir que la paciente presentó cambios descritos en publicaciones como el aumento de tamaño de las amígdalas y secreción nasal y los cambios iniciales del cuadro hemático que podrían o no presentarse o estar relacionados con otras patologías 3-11,13-15.
Sin embargo, otros signos descritos en el caso (como los cambios estructurales en el cartílago nasal, la depresión central en la punta de la nariz, la afectación de los cornetes, sonidos respiratorios superiores y disnea) no se han descrito en las publicaciones existentes y no es posible establecer la relación de estos signos con el SARS-CoV-2. Por lo anterior, es necesario adelantar más estudios que permitan profundizar esta información en animales, sobre todo en felinos silvestres. Cabe resaltar que una de las teorías iniciales (al momento de la presentación de la depresión central en la punta de la nariz y la observación de la destrucción de los cartílagos nasales en la radiografía, luego de descartar el origen traumático) fue el posible acenso de bacterias de cavidad oral que llegaron al cartílago nasal, provocando esta destrucción 27,28. Cabe mencionar que durante el desarrollo del caso se realizó un ELISA para la detección de coronavirus felino (FeCoV) y esta salió negativa, como sucedió en ocasiones anteriores a lo largo de su vida, como se evidenció en su historia clínica.
Se logró la identificación molecular del virus de SARS-CoV-2 en esta paciente, a partir de muestras de hisopados nasales, con la técnica utilizada para el diagnóstico en humanos (descrita en la metodología) por el laboratorio de Microbiología de la Facultad de Medicina de la Universidad Nacional de Colombia. Esto muestra, por un lado, que los protocolos y técnicas de diagnóstico para SARS-CoV-2 desarrolladas en humanos son aplicables en animales domésticos y silvestres como lo muestran los pocos estudios existentes 7-9,11,29-30 y en el caso colombiano, específicamente, es fundamental realizar más estudios para estandarizar el diagnóstico en animales y generar acceso a estos procesos desde la salud animal.
Con respecto a la secuenciación genómica, el hecho de que con la base de las pipetas utilizadas no pudiera obtener una alineación factible cuando se usó el genoma de FIP como referencia (pese a que pudo obtenerla cuando se usó NC_045512 como referencia) sugiere que el ARN muestreado está más cerca del genoma de referencia de Wuhan que del coronavirus causante de FIP.
Esta sugerencia también es compatible con la mayor similitud entre la región del gen codificante Spike del genoma ensamblado entre NCJ045512.2 y el genoma ensamblado, así como la ausencia total de similitud entre los genes E y M, entre NCJ045512.2 y el genoma ensamblado de Leona, en comparación al genoma de FIP.
La baja similitud general entre NCJ045512 y el genoma ensamblado, que también mostró un patrón cíclico de alineación seguido de una desalineación de principio a fin en ambos genomas, no permitió evaluar qué tan relacionados estaban ambos genomas. Por ello, se sospecha que una combinación de varios factores, como cebado, cobertura y mutaciones, son la causa de este patrón de ensamblaje.
Sin embargo, el genoma secuenciado por la presente fue clasificado como perteneciente al clado 20C del software Nextstrain, particularmente debido a las sustituciones identificadas P681H y D950N.
Por todo lo anterior, es preciso adelantar más investigaciones para entender las dinámicas de transmisión y patogénesis de esta infección en los animales domésticos y silvestres, y el rol que juegan para el entendimiento y la transmisión de este virus a los humanos 31-39 ♦

