











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
Permalink
Introducción
Los tumores inflamatorios miofibroblásticos son tumores raros, de etiología desconocida. Abarcan un espectro de proliferación miofibroblástica con una cantidad variable de infiltrado inflamatorio. Se ha denominado de diferentes maneras como pseudotumor inflamatorio, xantoma fibroso, granuloma de células plasmáticas, pseudosarcoma, hamartoma linfoide y, actualmente, como tumor inflamatorio miofibroblástico. Esta amplia nomenclatura refleja la incertidumbre respecto a su naturaleza 1.
Se han propuesto varias teorías respecto a su etiología, como un proceso reactivo a infecciones, procesos autoinmunes o síndrome paraneoplásico, pero la etiología continúa siendo desconocida. Se ha relacionado también con procesos infecciosos, metástasis y alteraciones genéticas cromosómicas clonales adquiridas 1.
Su presentación clínica puede ser muy variable, dependiendo del lugar anatómico donde se origine, y puede incluir sensación de masa, diaforesis, fiebre, pérdida de peso y dolor 2. Afecta principalmente niños y adultos jóvenes, pero puede aparecer en cualquier edad y no tiene preferencia por género alguno 3.
El diagnóstico histológico consiste en la proliferación de miofibroblastos mezclados con células inflamatorias, predominantemente mononucleares. Se requiere estudio de inmunohistoquímica, que muestra que las células tumorales son característicamente positivas para la actina del músculo liso, con o sin expresión de desmina; focalmente positivo para vimentina y negativo para CD117 y CD34 3,4. La quinasa del linfoma anaplásico, también conocida como receptor de tirosina quinasa ALK o CD246 ayuda a establecer el diagnóstico, que no se puede realizar mediante estudios de imágenes. El diagnóstico definitivo es histopatológico 4.
Inicialmente se consideraba que el pulmón era el órgano más comúnmente comprometido, sin embargo, publicaciones recientes muestran mayor incidencia en focos extrapulmonares como la cavidad abdominopélvica, siendo el hígado uno de los más comprometidos 5. Se han informado sitios atípicos como intestino, páncreas, aparato genitourinario y sistema óseo 6.
El tratamiento definitivo es la resección quirúrgica completa, aunque en algunos casos esta entidad tiene regresión espontánea, por lo que los autores han planteado la posibilidad del manejo conservador 5,7. Hasta la fecha, hay poca evidencia del papel de la quimioterapia en los tumores inflamatorios miofibroblásticos y la mayoría de los reportes corresponden a la población pediátrica 8. La radioterapia ha mostrado algunos beneficios en los tumores inflamatorios miofibroblásticos pulmonares, pero no parece tener beneficio en las localizaciones extrapulmonares 1.
La tasa de recurrencia varía según el sitio anatómico, desde 2 % en el pulmón hasta un 25 % en lesiones extrapulmonares 9.
Caso clínico
Paciente masculino de 67 años, con único antecedente de hipertensión arterial, que consultó por un cuadro clínico de dos meses de evolución de fiebre intermitente de predominio nocturno, con sensación de masa abdominal dolorosa en epigastrio; al interrogatorio negaba cambios en el hábito intestinal, náuseas, vómito o algún otro síntoma gastrointestinal. Como estudio inicial se realizó una tomografía computarizada contrastada de abdomen y pelvis en donde se describió una lesión de aspecto infiltrante tumoral primario, con compromiso de la grasa a nivel retroperitoneal en la transcavidad de los epiplones, y enfermedad diverticular (Figura 1).

Figura 1. Tomografía computarizada de abdomen y pelvis contrastada, las flechas señalan la masa sólida heterogénea compatible con un tumor miofibroblástico inflamatorio gastrointestinal. A. corte axial. B. corte coronal.
Se tomó biopsia percutánea de la lesión con hallazgos patológicos de pseudotumor inflamatorio. Por su compromiso en la cara posterior del estómago y mesocolon transverso se realizó una gastrectomía subtotal radical con reconstrucción en Y de Roux y colectomía parcial del transverso con anastomosis mecánica colo-colónica por laparotomía (Figura 2).

Figura 2. A. hallazgo intraoperatorio en bloque con el colon. B. espécimen quirúrgico del tumor miofibroblástico inflamatorio.
El resultado de la patología quirúrgica final informó tumor miofibroblástico inflamatorio, de compromiso multifocal, adherido a la serosa de estómago e intestino, sin compromiso muscular, bordes de resección libres de tumor, con 20 ganglios negativos para malignidad y epiplón normal (Figura 3). Los marcadores de inmunohistoquímica fueron positivos para CD68 en abundantes histiocitos y Desmina positiva en fibras musculares y fueron negativos para AML, CK, CD10, H Cladesmon, CD35, CD68, CD21, MDM2 ALK, S100, ki67 del 30 %.
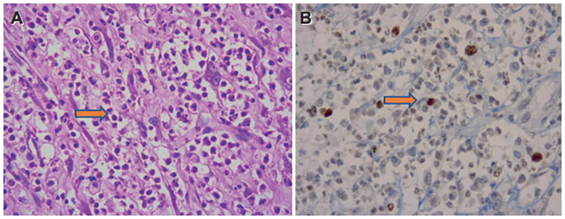
Discusión
El tumor miofibroblástico es una entidad que se agrupa dentro de los pseudotumores inflamatorios. Puede aparecer en cualquier grupo de edad, pero es más frecuente en las primeras dos décadas de la vida, con un promedio de edad al diagnóstico de 10 años, sin diferencia significativa entre los dos sexos 8.
La teoría de origen más aceptada es una respuesta inflamatoria exagerada posterior a un trauma localizado. En segundo lugar, se propone la infección como antecedente, la cual es producida con mayor frecuencia por micobacterias, virus de Epstein-Barr, Actinomyces y Mycoplasma. Se han demostrado otras asociaciones con Mycobacterium avium-complejo intracelular, Corynebacterium equi, Escherichia coli, Klebsiella, Bacillus sphaericus, Pseudomonas, Helicobacter pylori, Coxiella burnetii, e incluso virus de inmunodeficiencia humana. La etiología infecciosa es soportada por el papel de las citocinas, principalmente la interleucina 6 (IL-6), de la que se ha descrito una posible aproximación terapéutica específica 5.
Por su presentación clínica e imagenológica y por sus componentes histológicos variables, su diagnóstico es difícil. Se han identificado asociaciones con anormalidades cromosómicas, como translocaciones en el brazo largo del cromosoma 2 y en el brazo corto del cromosoma 9, y aberraciones citogenéticas, tales como el gen ALK (anaplastic lymphoma kinase) mutación p53 y expresión MDM2, lo cual podría, posteriormente, reconocerse como una lesión neoplásica específica o un verdadero tumor 6.
Las lesiones tumorales son únicas, pero pueden ser múltiples hasta en el 5 % de los casos. Se manifiesta clínicamente con signos y síntomas constitucionales dependiendo del sitio de origen y del efecto de masa sobre los órganos vecinos 9.
Conclusiones
El tumor miofibroblástico está catalogado como un tumor de comportamiento intermedio. Sus manifestaciones clínicas son variadas y están determinadas por el sitio anatómico afectado. El manejo depende de su localización, expresión de ALK, comportamiento y factibilidad de resección quirúrgica.
En este reporte se presentó el caso de un hombre adulto mayor con un tumor inflamatorio gastrointestinal, de compromiso multifocal, adherido a la serosa del estómago e intestino, sin compromiso muscular, manejado con cirugía radical, sin identificar malignidad, pocas veces descrito en la literatura.

