











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink1. Introduction
Cutinases are serine esterases that belong to the α/β hydrolase superfamily. They possess a classical Ser-His-Asp catalytic triad in which the catalytic serine is exposed to solvent because cutinases lack a hydrophobic lid that covers the serine active site in lipases. The cutinase active site is large enough to accommodate high molecular-weight substrates such as cutin and some synthetic polyesters as well as soluble low molecular weight esters and short and long chain triacylglycerols [1].
Cutinase is distinct among the α/β hydrolases because, unlike the majority of esterases [2], it can hydrolyze lipid substrates. Unlike lipases [3], its activity is not activated by interfacial effects, and its properties can be used to identify the cutinases [4].
Filamentous fungi are among the microorganisms that have the largest arsenal of cutinases. Given the diversity that exists of microorganisms with the potential to produce cutinases, it is necessary to have a practical method for their identification.
There are several methods to measure cutinolytic activity. The first method consisted of pH indicators in basic form with purified cutin as a carbon source [5]. Unfortunately, natural cutin is not commercially available, and it must be prepared by individual laboratories via extraction with organic solvent, followed by treatment with pectinases, cellulases, and lipases to eliminate contaminants. Its performance is minimal [6]. This may lead to errors in the selection of cutinase-producing microorganisms based on the reproducibility of the cutin preparation.
Other methods are based on rapid tests to rule out other hydrolytic enzymes such as esterases and lipases. From these, the most accepted is the rhodamine B method in conjunction with methods that use 4-nitrophenyl esters [7]. Initially the strains of interest are evaluated with short chain triglycerides in the presence of Rhodamine B showing a fluorescent halo in response. Here, the ester bond can only be cleaved by esterases and cutinases thus ruling out lipases that prefer long chains and a water-oil interface [3]. The microorganisms pre-selected in the previous stage should be inoculated in liquid mineral medium and incubated in the presence of cutin to induce the production of cutinases and finally analyze the supernatants with 4-nitrophenyl esters [6]. One limitation of this method is the growth of the strains with cutinase potential. The 4-nitrophenyl esters are suitable substrates, but they are highly susceptible to impurities in the preparation of the enzyme. Thus, if one does not select the right ester, then many lipases or esterases can hydrolyze them.
Methods of identification of cutinase in SDS-PAGE gels have also been developed where proteins can be renatured in a buffer solution, and their hydrolytic capacity evaluated in cutin via infrared spectroscopy [8]. However, this again leads to problems in preparation of the cutin. The difference is that the amount needed for the tests is only a few milligrams.
The following study compares screening methods that allow one to select cutinase-producing microorganisms with flaxseed oil as a substitute; this only uses cutin as a confirmatory test at the end of the process. This is due to the diversity of existing methods for the identification of cutinase-producing microorganisms that involve the preparation of cutin. This work is also based on studies showing that flaxseed oil is a better inducer of cutinases than cutin [9].
2. Materials and Methods
2.1. Biological material
Here, 33 strains isolated from Banana rachis residues was identified morphologically and molecularly [10]. The strains are grouped into 9 genera of filamentous fungi.
2.2. Preparation of the inoculums
The inoculum was prepared using aliquots of 10 mL distilled water for every 3 petri dishes with potato dextrose agar (PDA) inoculated with the fungal strains after 72 h of growth. The inoculum assayed 6.2x106 spores/mL [11].
2.3. Selection of microorganisms with cutinolytic activity
2.3.1. Basic indicator method
The cutinases-selective medium was used for screening and testing strains of filamentous fungi with high potential cutinase. This work consisted of a modified Czapeck-Dox medium (3 g NaNO3, 1 g K2HPO4, 0.5 g KCl, 0.01 g FeSO4.7H2O and 17 g agar-agar in 1 L of distilled water). Flaxseed oil (1% w/v) served as the sole source of carbon, and phenol red was an indicator at pH 9. When the color became yellow, the test was confirmed to have cutinases [5].
The negative control was a glucose carbon source; all tests were done in triplicate and incubated at 25 ° C.
2.3.2. Rhodamine B method
Fungal strains were inoculated into Petri dishes containing triacetin agar (containing the colorant Rhodamine B) culture medium and incubated at 30 ° C for 8 days. The formation of a fluorescent halo around a colony during growth indicated the production of the enzyme [7]. The test was done in triplicate.
2.3.3. P-NPB method
All strains were inoculated in 10 mL of a culture medium composed of 0.06% NaNO3, 0.06% K2HPO4, 0.02% MgSO4, 0.02% KCl, 0.01% FeSO4.7H2O, at pH 7.2, plus 1% (w/v) flaxseed oil as the carbon source.
The cultures were incubated at 30 ° C and shaken at 100 rpm for 96 hours. After culture development, the tubes were centrifuged at 1070 x G at 10°C, for 15 minutes. The cutinase and lipase activities were measured in the supernatants [7]. In the negative control, glucose was used as the carbon source, and all tests were carried out in triplicate.
2.3.3.1. Cutinase Assay
Cutinase activity was determined spectrophotometrically following the hydrolysis of P-NPB at 405 nm. An aliquot (70 μL) of the culture supernatant was added to 3.43 mL of a reaction mixture having the following composition: 0.56 mL P-NPB dissolved in 50 mM phosphate buffer at pH 7.0 with 0.2% (N/P) Triton X-100 and 0.43 M in tetrahydrofuran. The reaction was monitored for 1 minute against a blank solution. One unit of cutinolytic activity was defined as the amount of cutinase required to release one micromole of p-nitrophenol in one minute under the specified conditions [12].
2.3.3.2. Lipase Assay
One milliliter of the culture supernatant was added to a reaction mixture containing 2 mL of 0.1 M phosphate buffer at pH 7.0 and 5 mL of an emulsion with composition (v/v): 75% arabic gum (7%) and 25% olive oil. The solution was incubated at 37°C for 30 minutes with shaking at 120 rpm. After incubation, the reaction was stopped by adding 15 mL of ethanol: acetone 1: 1 (v/v). The fatty acids were released and titrated with 0.05 M NaOH. One unit of lipase activity was defined as the amount of enzyme required to release a micromole of oleic acid in one minute per mL under specified conditions [13]. Fig. 1 shows the selection process for each method.

2.4. Proteins and cutinase determination
The amount of total proteins was determined by the Bradford method, and protein presence was confirmed by electrophoresis.
2.4.1. Bradford method
The dye reagent was prepared according to Bradford [14]. Coomassie brilliant blue G-250 (100 mg) was dissolved in 50 mL 95% ethanol. A volume of 100 mL of phosphoric acid (85% w/v) was added, and the solution was diluted to 1 L with deionized water and immediately filtered twice. The dye reagent was stored at 4°C and protected from light.
For protein determinations, 50 μL culture supernatants were taken, and 200 μL of the Bradford reagent was added. The measurement was carried out after 5 minutes against a blank of deionized water. All tests were performed in triplicate.
The total protein concentration present in the culture supernatants was determined for a calibration curve using serum albumin (BSA) as a protein standard. The absorbance was measured at 450 nm and 595 nm and the Abs595/Abs450 ratio was calculated.
2.4.2. Protein electrophoresis
Electrophoresis was performed in a MiniProtean II vertical chamber (Bio-Rad Laboratories Inc., CA-USA), and molecular weight determinations were carried out with SDS-PAGE using 12% acrylamide gels [8].
The samples were mixed in a 3:1 ratio with 5X loading buffer (60 mM tris-HCl, 25% v/v glycerol, 2% w/v SDS, 14.4 mM 2-mercaptoethanol, and 0.1% bromophenol blue at pH 6.8). These were heated at 100 ° C for 5 minutes and then loaded on acrylamide gel. The electrophoresis was performed at a constant voltage of 200 V. Tris-glycine-SDS run buffer (tris 25 mM, glycine 192 mM and SDS 0.1% w/v) was used. The molecular weight of the proteins was determined by comparing their mobility with that of a mixture of proteins that varied in size from 15 kDa to 250 kDa.
After separating the protein samples by SDS-PAGE, the cutinase activity was determined using tomato cutin. The enzymes were renatured by washing the acrylamide gel with 0.1 M phosphate at pH 7.5 for 30 minutes with constant stirring at room temperature. A second wash was performed in the same phosphate buffer containing 5% Triton X-100 and the initial wash step was repeated.
2.5. Cutin extraction
Ripe tomatoes were purchased from the local market and were used to extract cutin. The tomatoes were peeled, and the peel added to a boiling oxalate buffer solution at pH 3.5 for 15 minutes or until it fully devoid of pulp. The peel was then filtered, washed, dried at 40°C, and extracted with chloroform-methanol (1: 1) and vacuum-dried. The resulting powder was treated with cellulase and pectinase in 0.05 M acetate buffer at pH 4.0 for 16 hours at room temperature with agitation, and finally filtered, washed, and dried at 40°C. The structure of the cutin was confirmed by FTIR.
2.6. Cutinases Identification
To identify cutinases, the renatured enzymes of the electrophoresis were reacted in the presence of tomato cutin dissolved in toluene, and hydrolysis was monitored by FTIR for 72 hours.
The cutinase was diluted 2.5-fold in phosphate buffer 0.05 M at pH 7.5. 700 μL of these solutions were used for the reactions, and then 5 mg of cutin previously suspended in 300 μL of toluene was added to achieve a final reaction volume of 1 mL. A blank where cutinase was not added was also similarly evaluated. Reactions were incubated for 72 hours at 37 ° C and 100 rpm.
To identify the hydrolysis product, the organic phase was extracted with chloroform and suction filtration in a Büchner funnel through a 0.45 μm nylon membrane (Whatman). The solvent was removed by suction and the samples were analyzed by FTIR.
3. Results and Discussion
3.1. Basic pH Indicator Method
Table 1 shows that for the basic indicator method, the Fusarium fujikuroi (H2), Penicillum chrysogenum (H7), Penicillium solitum (H10), and Mucor fragilis (H16) species grew and exhibited yellow zones after 8 days in presence of the phenol red pH indicator. This is because the pH of the medium decreased when the fatty acids released from the hydrolysis of flaxseed oil were released.
Table 1 Screening for the Selection of Cutinase Producing Microorganisms
| Species | Isolated | rating phenol red | rating Rhodamine B | Cutinase activity (U/mL) | Lipase activity (U/mL) |
|---|---|---|---|---|---|
| Syncephalastrum racemosus | H23 | + | - | 13,7 ± 0,2 | 0,1 ± 0,0 |
| Fusarium sp | H1 | - | - | 0 | 0 |
| Fusarium fujikuroi | H2 | ++ | ++ | 33,5 ± 2,0 | 0,1 ± 0,1 |
| Fusarium sp | H4 | + | - | 3,5 ± 0,2 | 2,3 ± 0,2 |
| Fusarium fujikuroi | H9 | + | - | 2,4 ± 0,4 | 33,6 ± 0,8 |
| Fusarium sp | H11 | - | - | 0,2 ± 0,0 | 1 ± 0,1 |
| Fusarium fujikuroi | H14 | + | - | 1 ± 0,0 | 1,1 ± 0,0 |
| Fusarium oxysporum | H17 | + | - | 0,4 ± 0,1 | 12,9 ± 0,2 |
| Fusarium fujikuroi | H19 | + | - | 9,35 ± 2,50 | 0,30 ± 0,02 |
| Fusarium fujikuroi | H24 | ++ | ++ | 3,3 ± 0,6 | 9,7 ± 0,5 |
| Fusarium sp | H27 | - | - | 0,3 ± 0,1 | 0,2 ± 0,1 |
| Fusarium equiseti | H29 | + | - | 2,2 ± 0,5 | 15,7 ± 0,7 |
| Trichoderma harzianum | H5 | + | ++ | 23,0 ± 1.9 | 0,9 ± 0,1 |
| Trichoderma harzianum | H12 | + | - | 1,1 ± 0,2 | 10,0 ± 0,2 |
| Penicillium sp | H6 | + | ++ | 0,5 ± 0,1 | 58,9 ± 0,3 |
| Penicillium chrysogenum | H7 | ++ | ++ | 39,3 ± 7,2 | 0,1 ± 0,0 |
| Penicillium solitum | H10 | ++ | ++ | 2,6 ± 0,2 | 6,5 ± 0,0 |
| Penicillium citrium | H25 | + | - | 0,6 ± 0,1 | 69,9 ± 2,0 |
| Penicillium chrysogenum | H36 | ++ | ++ | 1,5 ± 0,5 | 13,6 ± 0,6 |
| Cladosporium pseudocladosporoides | H31 | - | - | 15,9 ± 1,0 | 0,9 ± 0,1 |
| Cladosporium clasdosporoides | H32 | - | - | 0 | 0 |
| Verticillium luteoalbum | H18 | + | - | 0,7 ± 0,2 | 16,4 ± 0,5 |
| Verticillium luteoalbum | H22 | - | - | 2,9 ± 0,8 | 7,9 ± 0,4 |
| Verticillium luteoalbum | H26 | + | - | 25,0 ± 4,8 | 0,1 ± 0,0 |
| Verticillium nigrescens | H28 | + | - | 2,4 ± 0,3 | 23,8 ± 2,5 |
| Verticillium luteoalbum | H35 | - | - | 2,1 ± 0,5 | 21,9 ± 3,2 |
| Mucor nidicola | H13 | - | - | 2,4 ± 0,1 | 1,0 ±0,1 |
| Mucor fragilis | H16 | ++ | - | 9,9 ± 0,1 | 4,9 ± 0,0 |
| Mucor fragilis | H20 | + | - | 9,5 ± 0,1 | 0 |
| Mucor fragilis | H30 | + | - | 4,2 ± 1,2 | 11,9 ± 0,4 |
| Mucor piriformis | H34 | - | - | 0 | 0 |
| Phoma sp. | H21 | - | - | 0,2 ± 0,0 | 14,5 ± 2,7 |
| Botrytis cinerea | H33 | - | - | 0,7 ± 0,2 | 5,8 ± 0,2 |
Symbols: ++ = growth of fungi strains and detectable color change on selective medium, + = growth of fungi strains but no detectable color change on selective medium, - = no growth
Source: The Authors.
In contrast to glucose, the production of hydrolytic enzymes was inhibited by not exhibiting a change of coloration in the medium, which excluded the possibility that the dye had an effect on the growth of the fungus and that the color change was due to the presence of other acid molecules (Fig. 2).
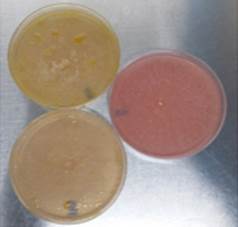
Source: The Authors.
Figure 2 Phenol red method: pink plate corresponds to glucose assay and yellow plates corresponds to flaxseed oil assay.
The test was convenient and rapid for the initial detection of cutinase production by filamentous fungi; 82% of the population were negative (33 strains x 82% discarded implies 6 confirmed strains). Thus, this is a slightly more selective alternative to 4-nitrophenyl esters. However, this method could be positive for lipases because these enzymes can cleave ester bonds of long chain triacylglycerols [3,15]. While this assay can be used to characterize the activity of cutinases, it cannot confirm that enzymes that produced the color change were cutinases.
3.2. Rhodamine B Method
Fig. 3 shows that due to the growth of fungi, the possible excreted hydrolytic enzymes released free acetic acid fatty acids from triacetin, which formed the quaternary ammonium salt [9- (2-carboxyphenyl)-6-diethylamine-3-xanthenylidene]-diethyl ammonium in the presence of Rhodamine B producing fluorescence. The Fusarium fujikuroi (H2), Penicillum chrysogenum (H7), Penicillium solitum (H10), Penicillum sp (H6) and Trichoderma harzarium (H5) species produced the fluorescent halo at 8 days of incubation (Table 1).

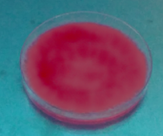
Source: The Authors.
Figure 3 Rhodamine B Method: first plate fluorescent halo, second plate initial condition.
Versus the previous method, Fusarium fujikuroi, Penicillum chrysogenum, and Penicillium solitum species could cleave ester bonds both long and short chain with and without a water-oil interface. This confirms the cutinase behavior [16]. Penicillum sp. and Trichoderma harzarium species tested positive for Rhodamine B. However, the phenol red tests had no change in coloration although they showed growth. This indicated that although there might be cutinases, their production was lower.
Mucor fragilis species showed no growth in the presence of triacetin agar. This confirmed that the enzyme could hydrolyze the long chain triglycylglyceride with a water-oil interface but not the short chain. This data confirmed the characteristic behavior of lipase [17].
The Rhodamine B method cannot definitively identify cutinases because estereses can also be positive. This type of assay can only pre-select cutinase-producing microorganisms, but their identity must be demonstrated by additional tests. For example, this cleaves the ester bonds in long-chain triacylglycerols. Thus, this method is a good complement to the phenol red method.
3.3. Cutinase assay with P-NPB
All strains were measured for activity with P-NPB. The most active species were: Fusarium fujikuroi (H2), Penicillum chrysogenum (H7), Verticillium leteoalbum (H26), Trichoderma harzianum (H5) and Cladosporium pseudocladosporoides (H31). These had values of 33.5 U/mL, 39.4 U/mL, 25 U/mL, 23 U/mL and 15.9 U/mL, respectively. Species Penicillium solitum (H10), Penicillum sp (H6) showed activities less than 5 U/mL.
Due to presence of a water-oil interface during incubation of the strains, the lipolytic activity of supernatants was measured to rule out a possible induction of lipases [18]. Penicillium solitum and Penicillium sp. species showed activities of 6.5 U/mL and 58 U/mL, respectively.
When comparing the results by the method of phenol red and Rhodamine B in the case of Penicillium solitum, a contribution to the decrease in pH could be produced by the generation of cutinases and lipases in the first method. In the second method, the fluorescent halo could only be developed via cutinases and esterases strongly induced by the substrate.
Penicillium sp showed a lipase activity of 58.9U/mL indicating a strong predisposition to produce lipases. However, the phenol red method was not strongly affected by these enzymes possibly due to a lack of good water-oil ratios that would induce their production. Activity with P-NPB for this species was 0.5 U/mL. The Rhodamine B assay formed a fluorescent halo. The reason that these two methods did not correlate could be the fact that the species was not a producer of cutinase but of esterase-induced by triacetin. The same could happen with Penicillum chrysogenum (H36) and Fusarium fujikuroi (H24).
Verticillium leteoalbum (H26) was positive for the phenol red method and negative for the Rhodamine B method indicating that the species was a producer of lipase and not of cutinases or esterases. However, the lipolytic activity was 0.9U/mL while the activity of P-NPB was 15.9 U/mL. The lack of concordance of the results could be because the species needs a mineral medium in addition to the substrate to produce cutinases; this mineral medium was absent in the Rhodamine B assay. This type of behavior is not usual for cutinases but there are studies that have shown that some cutinases require the presence of minerals to change the conformation of their active site. This type of cutinases they a hydrophobic cover and are called lipolytic cutinases [16].
Cladosporium pseudocladosporoides (H 31 ) showed an activity of 15.9 U/mL with P-PNB; the methods of phenol red and Rhodamine B were negative. This could be because the growth and enzymatic production was influenced by incubation temperature, humidity, and time, which led to the formation of isoforms dependent on the fermentation medium and therefore the components in the liquid medium stabilized the enzymes differently to give a positive response to the P-NPB assay [19,20].
Fusarium fujikuroi (H2), Penicillum chrysogenum (H7), and Trichoderma harzianum (H5) had results in the three methods that were concordant. The pH decreases in phenol red causing a change in the medium color and a fluorescent halo with Rhodamine B. This shows that they could splitting short and long chain triacylglycerol ester bonds. These data confirmed that the activity of P-PNB is greater than 20 U/mL with low lipase activity.
3.4. Cutinases Identification
Fig. 4 shows the specific activity of crude extracts from the 5 species selected by the P-NPB method. The species Fusarium fujikuro and Penicillum chrysogenum had the highest specific activity with activity values of 16.8 U/mg and 8.6 U/mg, respectively. Trichoderma harzianum had the highest protein value of 40.9 mg/mL. This could be because the amount of protein obtained did not correspond to cutinases as in previous cases.

Source: The Authors.
Figure 4 Comparison between total proteins, cutinase activity, and specific activity.
Fig. 5 shows the determination of the molecular weight of cutinases by SDS-PAGE electrophoresis. The analysis of the electrophoresis of the crude extracts shows proteins of low molecular weight (around 20-25 kDa) similar to those reported in the literature for fungal strains [21].

Source: The Authors.
Figure 5 Electrophoresis SDS-PAGE. Line 1 H26, line 2 molecular marker, line 3 H5, line 4 H31, line 5 H7 and line 6 H2.
Fig. 6 shows the spectra of the hydrolysis reaction between the crude extracts of species Fusarium fujikuro, Penicillum chrysogenum, and the tomato cutin via FTIR. Hydrolyzed cutin was obtained after 72 hours. The appearance of the carboxylic acid absorption bands can be observed in 1792 and 1770 cm-1 for H2 and H7, respectively, while an ester band at 1784 cm-1 is observed for cutin. In addition, the typical bands for the hydroxy group 3278 cm-1 were higher in the hydrolyzed cutin (3322-3378 cm-1), which explains why the hydroxyl groups were generated after hydrolysis of the cutin ester.

The main products due to the degradation of tomato cutin are 8, 16, 9, 16, 10, 16-dimethoxy-hexadecanoic acid, hydroxy- methoxy hexadecanoic acid, and palmitic acid derivatives. Fig. 6 shows the palmitic acid FTIR bands, which are similar to those obtained by the cutin hydrolyzed by Fusarium fujikuro and Penicillum chrysogenum confirming its degradation. The results are comparable to those mentioned in the literature for apple cutin and its hydrolysis [8].
Finally, the SDS-PAGE and FTIR suggests that the P-NPB method using flaxseed oil as an inducer can indicate if a strain or species is a producer of cutinases.
In conclusion, this study indicates that the P-NPB method with flaxseed oil as a substitute for cutin is a screening test to identify strains with activity cutinase. This step is more efficient than the phenol red and Rhodamine B methods based on a higher selectivity with the active site of the cutinases due to the use of P-NPB. It does not have difficulties associated with the preparation of the substrate. It also eliminates the disadvantages whereby lipases are an interferent.

