











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
1. Introduction
By definition, a bone defect is a lack of bone tissue in the body, where it should normally be. This could be caused by several congenital or acquired conditions, such as trauma, tumor resection or infections. The objective of bone regeneration is to use a material that acts as a template to guide bone growth and repair [1]. Although the use of autologous bone graft and bone allograft are the two most common options for bone regeneration [2], their clinical benefits are still not guaranteed. This is because complications and morbidity are frequently found in patients. Therefore, research on alternative bone substitutes is still important and necessary [3], and synthetic bone substitutes are still considered for bone tissue regeneration. Synthetic bone substitutes are used because of their biocompatibility and osteoconductivity, and the potential they provide to overcome the previously mentioned limitations of autologous and allogenic bone grafts.
Bone is a porous composite material of ceramic and polymeric parts, apatite and collagen respectively, with characteristic properties of strength, toughness, and controlled biodegradability. Additionally, it has biological properties such as biocompatibility, osteogenicity, osteoinductivity, and osteoconductivity. Therefore, a material designed to replace bone must possess certain characteristics. These include the following: the material must be porous (≈ 60 - 90% porosity of 100-500 µm in size); match the mechanical properties of the cortical bone (≈ 50 MPa of compressive strength, and 5 - 6 GPa of elastic modulus); and be able to be cut in a determined shape and pressed into the bone defect [1]. The interconnected porous structure guarantees fluid flow, vascularization, migration of cells, and bone ingrowth. Therefore, a bone substitute must exhibit an interconnected porous structure, which is the main limiting factor for osteoconductivity [4]. The cells that must migrate to the bone substitute are mesenchymal stem cells, osteoblasts, and osteocytes. The osteoinductivity of the material refers to the stimulation of the mesenchymal stem cells to differentiate in preosteoblasts and begin the bone formation process [5].
Calcium phosphates, calcium sulfates, and bioactive glasses are the most common materials clinically used for this application. Other materials used are collagen, growth factors and demineralized bone matrix [6]. Calcium phosphates exhibit similar structure and chemical composition to natural bone, which facilitates osteointegration with the host bone [7]. Calcium sulfate exhibits rapid resorption and ability to stimulate osteogenesis, as well as osteoinductivity [8]. The success of bioglasses is attributed to a hydroxycarbonate apatite layer formed on their surfaces when implanted, following initial glass dissolution. This layer interacts with collagen fibrils to integrate with the host bone. The bone matrix produced by the osteogenic cells is stimulated by the dissolution products of the bioglass [1].
The materials mentioned above exhibit some disadvantages, including reduced mechanical strength, fracture toughness and fatigue resistance [9]. The low values for these properties are because of the brittle nature of calcium phosphates, calcium sulfates, and bioglasses. These poor mechanical properties are increased in porous structures, such as scaffolds, which sometimes makes it impossible to use these materials in repairing large osseous defects. One of used strategies used to improve the mechanical behavior of calcium phosphates is to add bioceramics with better mechanical properties, e.g. zirconia ZrO2 [10], titania TiO2 [11], and alumina Al2O3 [11]. In general, these ceramics improve mechanical properties. This is the case with alumina, which early studies have shown to improve crack resistance through crack deflection, and to increase fracture toughness of phosphates [12-14]. However, these ceramics are nearly inert in the physiological environment [15]. It has been found, for example, that alumina diminishes the bioactivity of calcium phosphates [16]. Moreover, when alumina is added to HA, decomposition to tricalcium phosphate (TCP) is promoted, which decreases the mechanical properties [17]. It also has been considered that the decomposition of HA defines the percentage of porosity [10].
The addition of other ceramics, such as calcium titanate, has proved to be beneficial to the mechanical properties and bioactivity of calcium phosphates. In contrast to the effect of alumina, which diminishes HA decomposition, calcium titanate improves hardness, and fracture toughness can be controlled [18]. It has been found that this ceramic can spontaneously form apatite on its surface after soaking in SBF [19], which indicates its bioactivity [20]. Moreover, it also has been proved that calcium titanate can improve the bioactivity of calcium phosphates when used as a hard coating [21].
Therefore, the aim of this work was to manufacture a bio-ceramic material exhibiting high fracture toughness and bioactivity, using a mix of 50%Vol alumina, 37.5%Vol calcium titanate, and 12.5%Vol hydroxyapatite. Moreover, considering the importance of porosity on bio-ceramics, the effect of adding magnesium stearate as a porogen agent in crystalline phases formed after the sintering process was analyzed. Bioactivity after immersion in Hank’s solution were evaluated. The biomimetic growing patterns of bone-like apatite on the surface were also analyzed.
2. Materials and methods
The raw materials used for making the compounds were hydroxyapatite (HA) powder (Strem Chemicals - 99.9 % purity), calcium titanate (CT) (Super Conductor Materials Inc. - 99.9 % purity), and α-alumina (Leco, 99.0 % purity, with traces of Na2O). The incorporation of bubbles was achieved by the addition of magnesium stearate (MS) (from a local market) as the evaporation compound. These raw materials were characterized by laser granulometry (Hydro 2000MU (A)), using water as dispersant, to assess their particle size distribution; scanning electron microscopy (SEM, JEOL JSM-649 OLV) to evaluate the morphology of the powders; and XRD (Brucker D8 Discover), with a Cu target, to identify the crystalline phases of the materials.
The proportion of the materials in the compound was set at 12.5%Vol of HA, 37.5%Vol of CT, and 50%Vol of alumina. The addition of magnesium stearate was varied between 0, 5, 10 and 15%, to evaluate the effect of its addition on the resultant porosity, crystalline phases, toughness, and bioactivity. The mixing of the materials was done manually for half an hour for each sample.
Cylindrical samples of the compounds of 6 mm height and 12.7 mm diameter were made by pressing in a 50 Ton Enerpac uniaxial hydraulic press, using dies and H13 steel punches. The applied load was 8000 lbf, equivalent to 110 MPa of pressure. Two drops of isopropyl alcohol were used as the binder agent. Next, the samples were sintered in a P480 Nabertherm furnace under an air atmosphere. The sintering temperature was set at 1250 °C for 3 hours. The heating rate was 5 °C/min.
The surface morphology of the sintered samples was analyzed by SEM (JEOL JSM-649 OLV), to evaluate the porosity produced by the process, as well as the topography. The resultant crystalline phases were also analyzed by XRD (Brucker D8 Discover), with a Cu target. The bioactivity of the samples was measured by immersion in Hanks’ balanced salt solution (Sigma-Aldrich, with Ca and Mg and without phenol red) for 12 and 24 hours, introducing the Petri dishes into a CO2 incubator (BINDER) at 37 °C. The surface was analyzed by SEM and glancing incidence XRD to study the calcium phosphates formed on the surface. GIXRD was carried out using a Brucker D8 Discover diffractometer with Cu-Kα radiation (λ = 1.5406 Å, 30kV and 16 mA) at grazing angle of 2°. X'pert HighScore Plus software was used to determine present phases, and Origin software was used to make the deconvolution of the peaks.
3. Results and discussions
3.1 Raw material characterization
The magnesium stearate exhibits a flaky morphology, with an average diameter of 17 μm, as can be seen in Table 1 and part b) of Fig. 1.


The XRD pattern also corroborates the nature of the material, with peaks in the region of low 2θ angles [22]. Meanwhile, the hydroxyapatite (Fig. 2) exhibits a rounded and agglomerate morphology, and a bimodal particle size distribution.

The average size of the particles is 27 μm (Table 1). The XRD pattern matches well to hexagonal P63/m Ca10(PO4)6(OH)2, corroborating the nature of the material as hydroxyapatite [23]. However, deconvoluted peaks, shown in Fig. 3, also indicate the presence of monoclinic p2 1 /b hydroxyapatite [24].

The calcium titanate (Table 1 and Fig. 4) and alumina powders (Table 1 and Fig. 5) also exhibit a bimodal particle size distribution. The calcium titanate powder exhibits a lower particle size, of 2 μm on average, while the alumina exhibits an average size of 18 μm. The XRD pattern of the calcium titanate corresponds to two phases: orthorhombic Pbnm [25], mainly, and cubic Pm 3 m [26].
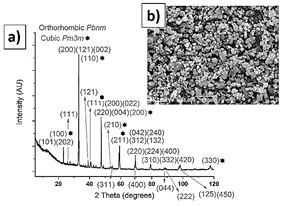
Source: The authors.
Figure 4 Calcium titanate XRD pattern, a); SEM morphology, b); and XRD pattern, c).

This is corroborated by the deconvoluted main peaks, shown in Fig. 6. The morphology of the alumina particles is agglomerated, and all the peaks exhibited by the alumina XRD pattern correspond to α-alumina [27,28]. However, due to the chemical composition, with traces of Na2O, convoluted peaks are observed (Fig. 7). The main peaks correspond to α-alumina, and the secondary peaks correspond to β-alumina, which is a solid solution of Na2O and Al2O3 [29].


3.2 Sintered material characterization
3.2.1 Crystalline phases
Fig. 8 shows the surface morphology of the sintered samples.

The sample with 0 % of MS used in the raw material does not exhibit pores in its surface. This is also the case of the sample with 5 % MS addition, although increased surface roughness is observed. The increase in roughness is beneficial, as the rougher surface is expected to promote wettability and, therefore, bioactivity. When 10 and 15 % of MS is added, some pores of several micrometers are observed. As expected, the sample with 15 % MS addition exhibits a higher porosity, due to the thermal decomposition of this material. At the micrometer scale, an alumina matrix is observed, with enriched regions of calcium titanate and calcium phosphates. This is shown in Fig. 9.

Source: The authors.
Figure 9 Micrograph of the 15%MS sample surface showing the phase distribution: zones with vertical arrows correspond to CT enriched zones, and zones with horizontal arrows correspond to CP enriched zones in the alumina matrix.
The XRD pattern of the sintered samples is shown in Fig. 10.
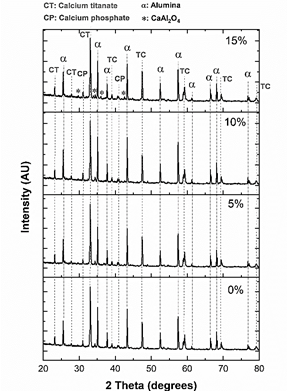
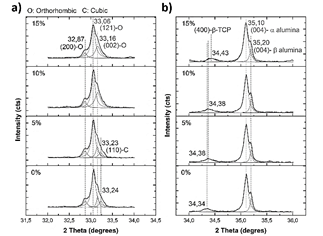
Samples are composed of α and β alumina, orthorhombic and cubic calcium titanate, oxyhydroxyapatite, β-tricalcium phosphate, and CaAl2O4. The main peak, which corresponds to calcium titanate, is located at 33° and was deconvoluted using lorentzian curves (Fig. 11- a)). Peaks of the samples with 0 and 5% of MS addition, as was the case with the CT powder, evidence the presence of orthorhombic and cubic phases of CT. However, when 10 and 15% of magnesium stearate was added, no presence of the cubic phase is evidenced. This means that the cubic phase disappeared at high contents of magnesium stearate. In the case of alumina (Fig. 11- b)), the presence of α and β phases is still observed, although the peaks have slightly shifted toward higher angles, indicating the presence of compressive stresses. A new peak located at 34.3° (Fig. 11- b)) is observed, which probably corresponds to (400) plane of β tricalcium phosphate (this is the second main peak of β-TCP) [30].
Source: The authors.
Figure 11 Calcium titanate, a), and alumina, b) main peaks of the XRD patterns after sintering.
In the case of calcium phosphate phases, it is important to understand the changes that HA experiences at high temperatures. When hydroxyapatite is heated to high temperatures and low water pressure, it experiences dehydration, and can produce oxyhydroxyapatites or oxyapatite, which can also decompose in tricalcium phosphate (TCP) and tetracalcium phosphate (TTCP). The removal of water implies the loss of OH groups of HA, changing from Ca5 (PO4 )3 (OH) to Ca10 (PO4 )6 O , which is oxyapatite. Depending on the degree of loss of OH groups, the resultant material can consist of a mix of HA and oxyapatite, called oxyhydroxyapatite (OHAP), and TCP and TTCP [31]. Additionally, when the material is cooled, part of TTCP and TCP directly reconstruct into OHAP by rehydration because of the presence of atmospheric water [32].
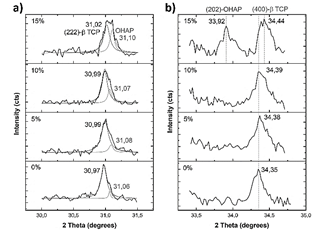
Therefore, shifting of the main peak, initially located at 31.7° in HA, towards lower angles, until reaching around 31.0°, indicates the transformation of HA. This peak can be deconvoluted in two different peaks, as can be seen in part a) of Fig. 12.
Source: The authors.
Figure 12 Calcium phosphates main peaks of the XRD patterns after sintering. Peak located around 31°, a); and peak located around 34.3°, b).
The peak located at lower angles corresponds to the main peak of β-TCP, while the peak located at higher angles apparently corresponds to OHAP. This is corroborated by the appearance of a peak located at 33.9° in the sample sintered with 15% of magnesium stearate, which corresponds to (202) plane of OHAP [32]. The greater presence of OHAP with the addition of magnesium stearate is evident. This greater presence of magnesium stearate implies a higher porosity in the samples, which in turn implies lower thermal conductivity and better permeability, both in heating and cooling. In cooling, the higher permeability benefits the diffusivity of the atmospheric water inside the material, propitiating the greater presence of OHAP phase at higher content of magnesium stearate in the raw material. The lower heat transfer and higher surface area also appear to promote the presence of orthorhombic rather than cubic phase in CT.
Small peaks located at 29.8°, 36.0° (in the sample sintered with 15% of MS), 41.7° and 42.6° (all samples) correspond to CaAl2O4, a compound between alumina and calcium [33], that has been previously observed in alumina-calcium phosphates compounds [34]. The shifting of the main peak of alumina toward higher angles with the addition of MS could indicate the dissolution of Ca in the alumina. The higher quantity of diffracted peaks in the 15%MS sample could indicate that the greater presence of pores promotes the formation of this phase, possibly due to the better diffusivity of ions caused by the higher exposure to heat.
The presence of crystalline phases in the sintered samples is summarized in Table 2.

3.2.2 Bioactivity and growing pattern
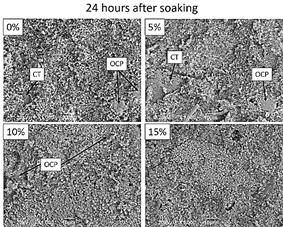
SEM images shown in Figs. 13 and 14 evidence a precipitation on the sample surfaces after 12 and 24 hours, respectively, of soaking in Hank’s solution. The crystal structure of calcium phosphate phases is confirmed with XRD (Fig. 15). Two phases are observed: triclinic octacalcium phosphate and hexagonal hydroxyapatite. The precipitate morphology is different in each sample and time. The 0%MS sample exhibits bigger precipitates of flaky shape morphology (related with the presence of octacalcium phosphate, OCP [35, 36]) after 12 hours of soaking. The presence of this type of precipitate diminishes at 24 hours. It is important to note the growth of HA on the OCP crystals, confirmed by the XRD pattern (Fig. 13- 0%). Fig. 14 shows the lower bioactivity of the enriched - CT regions in the 0%MS sample. This behavior is also observed in the 5%MS sample, but is observed to a lower extent in the 10 and 15%MS samples (Figs. 13 and 14). This could indicate that the cubic phase could act negatively in the bioactivity of calcium titanate.

Source: The authors.
Figure 13 SEM morphology of the surface of the sintered samples after 12 hours of immersion in Hank’s solution.
Source: The authors.
Figure 14 SEM morphology of the surface of the sintered samples after 24 hours of immersion in Hank’s solution.
From Figs. 13 and 14, it is also clear that the precipitates become smaller with the addition of MS in raw materials. Micrographs of the 10 and 15%MS samples after 24 hours of soaking evidence different growing patterns on the part of the phosphates. In the 15%MS samples, the growth of a high quantity of calcium phosphate seems to be directional and perpendicular to the surface. That is not the case for the 10%MS sample.
All diffraction peaks were sharp, indicating a high degree of crystallinity of the grown phosphate on the surface (Fig. 15). However, the ratio between the intensities of the main peaks implies different growth patterns of the calcium phosphates, depending on the porosity of the samples. This is explained next.
Source: The authors.
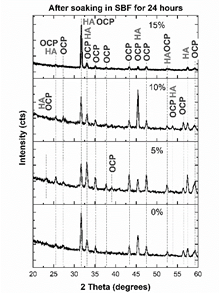
Figure 15 XRD pattern of the sample surface after 24 hours soaking in Hank’s solution.
Although HA is thermodynamically more stable than OCP, the rate of precipitation of HA in SBF is lower than that exhibited for OCP, which explains the presence of OCP in addition to HA in the samples. Moreover, some researchers have shown that OCP is the in-vivo precursor for the bone-like apatite formation. The transformation of OCP to HA is mediated by the bone-cells, which resorb phosphates and make bone [37]. OCP firstly precipitates, and then hydrolyzes to a transition product intermediate to OCP and HA [38-40]. It has been proved that, in in vitro biological conditions, dicalcium phosphate dehydrate (DCPD) is formed at first, but this rapidly transforms into OCP or HA [40]. OCP transforms to HA because they exhibit similar atomic arrangement, which provides favorable sites for HA nucleation [36]. The precipitation of OCP is positive, because it has been proved that this phosphate is more bioactive than HA, and it exhibits stimulatory capacity of osteoblastic differentiation in osteocytes [41].
Although the detection of HA growth in the presence of OCP is difficult because XRD patterns of HA and OCP are similar, the deconvolution of peaks can elucidate the difference. Therefore, Figs. 16 and 17 show the deconvoluted peaks.
Source: The authors.
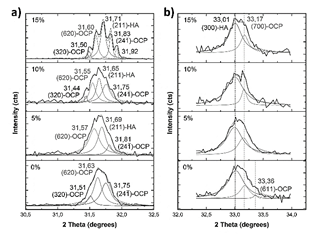
Figure 16 Deconvoluted main XRD peaks after soaking in Hank’s solution. Peak located around 31.7°, a); and peak located around 33.1°, b).
Source: The authors.
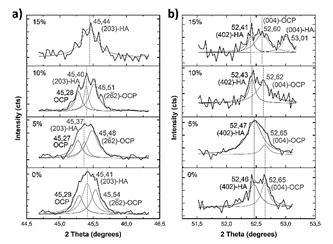
Figure 17 Deconvoluted XRD peaks after soaking. Peak located around 45.4°, a); and peak located around 52.5°, b).
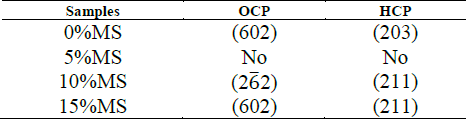
When MS is not added to the raw material of the compounds, the main peak can be deconvoluted in three peaks (Fig. 16 - a)): a peak located at 31.6° corresponding to (620) plane of OCP [43], and peaks located at 31.5° and 31.7° corresponding to (320) and (24 1 ) planes [40]. This means that the (211) main peak of HA is not diffracted in this sample. However, (300) plane of HA is diffracted at 33.1°, as (203) and (402) at 45.4 and 52.5° [44], respectively, as seen in Fig. 17. This corroborates the presence of these phosphates with a preferential direction growth in precipitation: (602) for OCP and (203) for HA.
When 5% of MS was added to the raw material of the compounds, the peaks located at 31° and 33° were also deconvoluted (Fig. 16). The (211) peak of HA appears diffracted, and no relevant preferred orientation of HA or OCP is observed. When 10% of MS was added to the raw material of the compounds, HA grows preferentially in (211) direction, while OCP grows preferentially in (2 6 2) plane [45]. When 15% of MS was added to the raw material of the compounds, (211) plane of HA is mainly diffracted, diminishing the intensity of OCP peaks. This corroborates the directional growth pattern of phosphates in Fig. 14 in this sample. However, although preferential direction growth is observed, this does not evidence an epitaxial growth, as was found in other studies [37,46,47]. Table 3 summarizes the preferential orientation of CP phases formed after soaking.
The transformation of OCP to HA is explained by Xin et al, through eq. (1), as a solid-state transformation, not by dissolution. During the transformation, side-products are generated as bubbles of H3PO4 and water [36].

It is important to note the appearance of a peak diffracted at 53.01° (Fig. 17-b) in sample with 15%MS, which corresponds to the (004) plane of HA. In addition to the great intensity of HA peaks, the appearance of this peak could indicate a higher quantity of HA in comparison to OCP in this sample. The higher intensity and appearance of OCP peaks at lower contents of MS used in the raw material could be related to the greater quantity of β-TCP, because it has been found that this phosphate favors the precipitation of OCP on its surface [43].
This crystal growth of HA along (211) direction is uncommon. Its preferential growth orientation is along the c-axis of hexagonal crystal structure. Growth along c-axis is referred to as c-plane, while growth along other directions are referred to as a,b-plane. Depending on this, HA exhibits different functions: a,b-plane HA exhibits bioactive functions, while c-plane HA shows more bio-inert functions. It has been found that a,b-plane HA is active in human saliva, while c-plane HA covers human teeth [48]. Therefore, in bone fillers a,b-plane HA could be more favorable [49].
3.2.3 Bioactivity and growing pattern
Based on previous research and the results of this study, we will now explain how each phase of the compounds interacts with simulated body fluids.
Biomaterials such as calcium phosphates interact with body fluids by dissolution, solubility, and precipitation [50]. The surface chemistry plays an important role in the calcium phosphates precipitation in SBF. The process depends on negative groups on the surfaces, which depend in turn on the negative ions present on the phosphates. Calcium ions are attracted to the surface, forming a positive layer, which will then attract negative ions [51]. In the case of HA, at the beginning of the interaction, the solubilization starts with the chemical interaction between OH groups and protons of the fluid, releasing water molecules and transforming to Ca3 (PO4 )2. Next, the solubilization of HA continues through the chemical interaction between phosphate groups and protons, which forms HPO4-2, interacting with and dissolving calcium. When HA has transformed to CaHPO4, equilibrium between fluid and modified HA is achieved, and the precipitation starts [51]. Oxy-hydroxyapatite is a partially de-hydroxylated HA, which means it is easier for this material to achieve equilibrium with the fluid, as is the case with β-TCP.
Although alumina also dissolves in SBF, it exhibits poorer bioactivity in comparison to HA due to its lower degradation rate; the weight loss of alumina in SBF is an order of magnitude less than that of the HA [52]. However, alumina contains a negative surface because of the presence of oxygen ions, which can attract calcium ions and form a positive layer, which will then attract negative ions. It has been found, for example, that alumina addition to some polymers can improve bioactivity and cell proliferation [53]. The growth of calcium phosphates on alumina matrix is evident in SEM photographs (Figs. 13 and 14).
In contrast, as expected, calcium titanate exhibited poor bioactivity. However, poor bioactivity CT zones are less frequently observed in samples with higher level of porosity, as shown in Fig. 14. As mentioned, this could indicate that orthorhombic phase favors bioactivity more than cubic phase. In the case of calcium phosphates, calcium ions are easily released to the medium, but that is not the case with calcium titanate, since it has a strong binding energy, restricting the release of Ca ions. This makes the bioactivity of this phase lower than that exhibited by calcium phosphates. On the other hand, the surface of calcium titanate is not as negative as the alumina surface, so it does not attract positive ions of the solutions onto its surface as easily.
The interaction of the three phases, which comprise the compounds alumina, calcium titanate, and calcium phosphates, with Hank’s solution is schematized in Fig. 18.

4. Conclusions
Porous compounds of alumina, calcium titanate, and calcium phosphates were successfully synthetized.
Due to the porosity, which causes lower heat transfer and higher permeability in the compounds during processing, the constituent materials can crystallize in different phases. Higher porosity causes higher presence of OHAP than β-TCP in calcium phosphates, inhibits the presence of cubic calcium titanate, and favors the presence of CaAl2O4.
At micrometer scale, an alumina matrix is observed, with enriched zones of calcium phosphates and calcium titanate. Calcium phosphates zones in the alumina matrix exhibited good bioactivity, in contrast to calcium titanate zones, which exhibited poor bioactivity.
Crystalline octacalcium phosphate and hydroxyapatite were formed on the surface of the materials after soaking by 24 hours in Hank’s solution. The relative intensities of the XRD peaks evidenced greater presence of OCP at low levels of porosity, and greater presence of HA at high levels of porosity.
(211) preferential growth direction of HA was observed at high level of porosity, especially in 15%MS sample, which is more favorable for bone filler applications.

