











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCTION
Leishmaniasis are neglected parasitic diseases caused by more than 20 protozoan species of the genus Leishmania, considered endemic in 98 countries [1]. Transmission to humans occurs during the blood repast of Phlebotomines from the Phlebotomus (Old World) and Lutzomyia (New World) families. Only the hematophagous females are responsible for the inoculation of promastigote forms in the host's skin [2]. The species Leishmania amazonensis is associated with the development of cutaneous leishmani-asis with the formation of necrotic ulcers, and is capable of causing the disseminated form of the disease [3].
Pharmacological therapy with pentavalent antimonials (N-methylglucamine antimoniate) is the first choice treatment, and amphotericin B and pentamidine are the second choice [4]. And there are successful reports of the use of miltefosine in cases of antimonium-resistant protozoa [5, 6]. However, the treatment of leishmaniasis is still a difficulty, considering that the available pharmacological therapies present limitations in terms of efficacy and safety, prolonging the treatment; therefore, a range of adverse reactions, the need for parenteral administration, in addition to the possible emergence of resistance, lead to low adherence to treatment by patients.
Several efforts search for bioactive natural compounds that can be used in the treatment of parasitic diseases [7]. Lapachol, α- and β-lapachones are promising natural naphthoquinones for Medicinal Chemistry due to their structural properties. Goulart et al. described the leishmanicidal activity of lapachol and some derivatives, showing the pharmacological potential of these substances [8]. In 2013, Guimarães et al. presented the activity oflapachol, α- and β-lapachones against the promastigote forms of four species of the genus Leishmania, ratifying the relevance of these naphthoquinones in the development of new leishmanicides [9].
In a study published this year, Souza et al. (2020) pointed out the relevance of the presence of isonicotinohydrazone and phthalazinylhydrazone nuclei in the structures of compounds for the antileishmanial activity. In this work, the authors synthesized five hydrazones and evaluated in vitro the activity against the promastigote form of L. amazonensis [10].
The development of new substances with therapeutic potential is a complex task involving multi and interdisciplinary efforts. Several strategies are used by Medicinal Chemistry uses to make the process of developing new drugs more effective. Molecular planning is one of the crucial steps in the process, and Molecular Hybridization (MH) is an effective alternative for the rational design of molecular structures of new prototype compounds. In this proposal, hybrid compounds are the result of joining molecular structures of distinct bioactive compounds [11, 12].
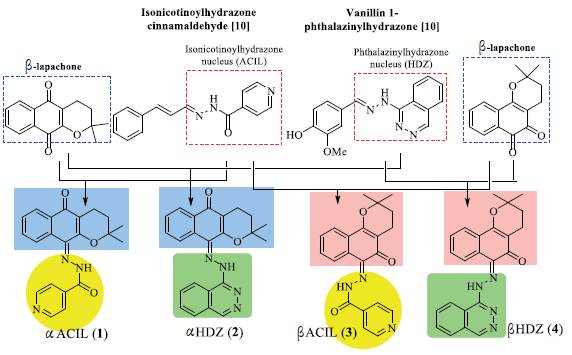
Considering the activity against protozoa of the genus Leishmania presented by the α-, β-lapachones and compounds containing the isonicotinoylhydrazone (ACIL) and phthalazinylhydrazone (HDZ) nuclei, four compounds were designer through the of MH strategy employing the molecular structures of the naphthoquinones and the ACIL and HDZ nuclei, as shown in figure 1. Subsequently, the hybrids were prepared and had their activities evaluated against the promastigote forms of L. amazonensis, L. infantum and L. major, besides the cytotoxic evaluation in murine macrophages.
MATERIALS AND METHODS
For the synthesis, all the reagents used were obtained from commercial sources and used without prior purification, with the exception of hydralazine hydrochloride, which was obtained from Apresolina* pills (Anovis Industrial Farmacêutica Ltda) [10, 13]. The synthesized substances had its melting temperatures determined in triplicate using analog fusiometer, model PFM-II (MS Tecnopon* instrumentation). The chromatographic profile of the substances was determined by Analytical Thin Layer Chromatography (TLC), using 2x4 cm aluminum/silica gel 60 plates with UV254 fluorescence indicator, revealed in ultraviolet (UV) or with iodine (I2). The purification in chromatographic column used silica gel 60 (70 - 230 mesh) and ethyl acetate/hexane (AcOEt/Hex) mixture with increasing polarity, as a mobile phase. After preparation and purification all, the synthesis products were stored under refrigeration and protected from light.
The structural elucidation of naphthhydrazones αACIL (1), αHDZ (2), βACIL (3) e βHDZ (4) was performed using 1H and 13C Nuclear Magnetic Resonance techniques (1H and 13C NMR) [13]. The NMR spectra have been registered in a Bruker Ascend TM 400 device, which operates at 400 MHz for the 1H nuclei and at 100 MHz for the 13C nuclei. The chemical displacements (δ) were given in ppm using tetramethylsilane solvent (TMS) as internal standard. All samples were solubilized in deuterated solvent (CDCl3 or DMSO-d6).
Infrared (IR) absorption spectra were obtained using PerkinElmer* (Spectrum 400) equipment with Attenuated Total Reflectance (ATR) device with zinc selenide crystal, from 4000 to 650 cm-1, and 4 cm resolution-1.
Extraction, purification, and characterization of lapachol
Lapachol was extracted from the stalk of the ipe (Tabebuia sp.). The splinters of the heartwood were submerged in an aqueous solution of sodium hydroxide (NaOH) at 1 % (m-v-1) for about 24 h. The filtrate was then acidified with a solution hydrochloric acid 6 M (HCl), and the lapachol was precipitated in the aqueous medium as a yellow solid. The solid was filtered by vacuum filtration and dried at room temperature.
Lapachol was purified by recrystallization from ethanol/water. Yellow crystalline solid, yield of 1.5 % (m/m) and melting point (m.p.): 138.33-140.33 °C. 1H NMR (400 MHz, CDCl3 , δ in ppm): 8.12 (1H, m, H5); 8.07 (1H, m, H8); 7.75 (1H, td,J = 7.6, 1.4 Hz, H6); 7.67 (1H, td, J = 7.5, 1.4 Hz, H7); 7.33 (1H, s, -OH); 5.21 (1H, m, H10); 3.31 (2H, d, J = 7.4 Hz, -CH2); 1.79 (3H, s, -CH3); 1.69 (3H, s, -CH3). 13C NMR (100 MHz, CDCL,, δ in ppm): 184.59 (C4); 181.70 (C1); 152.67 (C2); 134.87 (C6); 133.87 (C4 and C11); 132.88 (C7); 129.41 (C8); 126.77 (C5); 126.06 (C8); 123.45 (C3); 119.62 (C10); 25.77 (-CH3); 22.61 (C9); 17.90 (-CH3').
Synthesis, purification, and characterization of α-lapachone
Lapachol (1 mmol; 242 mg) was dissolved in a solution of glacial acetic acid (AcOH; 240 μL) and concentrated HCl (630 μL). It was heated to 100 °C for 1.5 h, and then cooled to room temperature. The reaction mixture was poured into a beaker containing cold distilled water, the precipitated pale yellow solid was iltered and dried at room temperature. The obtained solid was purified by simple recrystallization using ethanol. Yellow crystals, yield of 71 % and m.p.: 116.33-117.66 °C. 1H NMR (400 MHz, CDCL,, δ in ppm): 8.08 (2H, m, H6 and H9); 7.68 (2H, m, H7 and H8); 2.63 (2H, t, J = 6.6 Hz, H4); 1.83 (2H, t, J = 6.6 Hz, H3); 1.44 (6H, s, 2 -CH3). 13C NMR (100 MHz, CDCL,, δ in ppm): 184.42 (C5); 180.03 (C10); 154.62 (C1); 133.87 (C7); 132.93 (C8); 132.07 (C5↔C9); 131.16 (C9 ↔ C5); 126.33 (C6); 125.97 (C9); 120.15 (C4); 78.17 (C2); 31.41 (C3); 26.51 (2 -CH3); 16.74 (C4).
Synthesis, purification, and characterization of β-lapachone
Cooled concentrated sulfuric acid (H2SO4) (730 μL) was added to a reaction vessel (25 mL) containing lapachol (1 mmol; 242 mg) and immersed in an ice bath at 0 °C. The reaction was stirred for 20 min. Then, the reaction mixture was poured into a beaker containing cold distilled water, the precipitated orange solid was iltered and dried at room temperature. β-lapachone was puriied by column chromatography. Reddish-orange crystals, yield of 69 % and m.p.: 154.66-158.0 °C. 1H NMR (400 MHz, DMSO-d6, δ in ppm): 7.91 (1H, d, J = 7.6 Hz, H7); 7.77 (2H, m, H9 and H10); 7.61 (1H, m, H8); 2.40 (2H, t, J = 6.6 Hz, H4); 1.82 (2H, t, J = 6.6 Hz, H3); 1.43 (6H, s, 2-CH3). 13C NMR (100 MHz, DMSO-d6, δ in ppm): 179.06 (C6); 177.83 (C5); 160.65 (C1); 135.02 (C9); 132.10 (C10); 130.84 (C8); 129.96 (c6); 127.83 (C7); 123.70 (C10); 112.51 (C4); 79.07 (C2); 30.81 (C3); 26.33 (2 -CH3); 15.97 (C4).
General procedure for the preparation of the compounds αACIL (1) and βACIL (3)
To a methanolic solution (5 mL) containing isoniazid (90 mg, 0.6 mmol) and the appropriate naphthoquinone (121 mg, 0.5 mmol) (α- or β-lapachone) was added a drop of concentrated HCl (37 %). The reaction mixture was stirred until complete consumption of naphthoquinone. The crystals formed were filtered, washed with methanol and distilled water and dried at room temperature. The product was purified by column chromatography [13].
General procedure for the preparation of compounds αHDZ (2) and βHDZ (4)
In a solution containing hydralazine hydrochloride (1.5 mmol) in methanol (11 mL), H2SO4 concentrate (approximately 700 μL) was slowly added, followed by appropriate naphthoquinone (121 mg, 0.5 mmol) (α- or β-lapachone). After the end of the reaction, the mixture was neutralized with 5 % (m-v-1) NaHCO3 solution. The precipitate formed was filtered, washed with distilled water and dried at room temperature. The product was purified by column chromatography [13].
Data from αACIL (1): (Z)-N'-(2,2-dimethyl-5-oxo-3,4-dihydro-2H-benzo[g] chromen-10(5H)-ylidene)isonicotinohydrazide
The reaction mixture remained under agitation at room temperature for 72 h [13]. Greenish-yellow solid (figure 2), yield of11.3 % and m.p.: 226.50-227.83 °C. 1H NMR (400 MHz, CDCl3, δ in ppm): 12.80 (1H, s, N-H); 8.86 (1H, m, H16 and H18); 8.52 (1H, d, J = 7.9 Hz, H9); 8.11 (1H, m, H6); 7.74 (2H, dd, J = 4.4; 1.6 Hz, H15 and H19); 7.64 (1H, dd, J = 12.9; 4.6 Hz, H8); 7.54 (1H, t,J = 7.5 Hz, H7); 2.69 (2H, t, J = 6.1 Hz, H4); 1.93 (2H, t, J = 6.3 Hz, H3); 1.53 (6H, s, 2 -CH3). 13C NMR (100 MHz, CDCl3, δ in ppm): 183.43 (C5); 161.99 (C13); 154.51 (C1a); 150.94 (C16 and C18); 140.39 (C14); 134.93 (C10); 133.22 (C9a); 132.45 (C8); 129.84 (C7); 129.61 (C5a); 125.70 (C6); 124.93 (C9); 120.96 (C15 and C19); 117.33 (C4a); 79.91 (C2); 31.14 (C3); 26.97 (2 -CH3); 16.88 (C4). IR (ATR, v in cm-1): 3330 (N-H); 1697 (C=O; N-C=O); 1633 (C=N); 1605-1455 (C=CAr); 1265-1229 and 874 (C-O); 907-688 (C-HAr). HRMS (ES+) calculated for C21H19N3O3 [M+H]+: 362.1504. Found: 362.1469.

Data from αHDZ (2): (Z) -2.2-dimethyl-10-(2-(phthalazin-1-yl)hydrazone)-3.4-dihydro-2H-benzo[g]chromen-5(10H)-one
The reaction mixture remained under stirring and reflux for 6 h [13]. Red crystalline solid (figure 2), yield of 10.1 % and m.p.: 238.83-240.50 °C. 1H NMR (400 MHz, CDCl3, δ in ppm): 11.11 (1H, s, N-H); 9.75 (1H, d,J = 8.1 Hz, H9); 8.56 (1H, dd,J = 6.1; 3.2 Hz, H15); 8.27 (1H, dd,J = 7.8; 1.4 Hz, H6); 8.10 (1H, s, H20); 7.80 (2H, m, H16 and H17); 7.66 (2H, m, H8 and H18); 7.53 (1H, m, H7); 2.66 (2H, t,J = 6.7 Hz, H4); 1.84 (2H, t, J = 6.7 Hz, H3); 1.46 (6H, s, 2 -CH3). 13C NMR (100 MHz, CDCl3, δ in ppm): 184.37 (C5); 159.37 (C1a); 150.96 (C13); 141.73 (C10); 140.13 (C20); 133.06 (C17); 132.39 (C16); 131.59 (C8); 130.90 (C9); 130.83 (C5a); 129.33 (C7); 128.89 (C9a); 127.82 (C19↔C14); 126.71 (C14 ↔ C19); 126.41 (C18); 126.11 (C6); 125.34 (C15); 113.51 (C4a); 77.36 (C2); 31.75 (C3); 26.74 (2 -CH3); 17.09 (C4). IR (ATR, v in cm-1): 3346 (N-H); 1622 (C=O; C=N); 1598 and 1572 (C=N; -C=N-N=C-); 1488, 1448 and 1396 (C=CAr); 1368 and 1350 (C-N); 1246-1231 and 865 (C-O); 907-691 (C-HAr). HRMS (eS+) calculated for C23H20N4O2 [M+H]+: 385.1664. Found: 385.1657.
Data from βACIL (3): (E)-N'-(2,2-dimethyl-5-oxo-3,4-dihydro-2H-benzo[h] chromen-6(5H)-ylidene)isonicotinohydrazide
The reaction mixture was stirred at room temperature for 10 minutes [13]. Yellow-orange solid (figure 2), yield of 66 % and m.p.: 237.66-238.50 °C. 1H NMR (400 MHz, CDCl3, δ in ppm): 8.86 (2H, d, J = 5.8 Hz, H16 and H18); 8.46 (1H, s, -NH); 7.89 (4H, m, 7H7, H10, H15 and H19); 7.50 (2H, m, H8 and H9); 2.61 (2H, t, J = 6.6 Hz, H4); 1.90 (2H,t, J = 6.7 Hz, H3); 1.49 (6H, s, 2 -CH3). 13C NMR (100 MHz, CDCl3, δ in ppm): 182.29 (C5); 163.38 (C1a and C13); 150.76 (C16 and C18); 139.67 (C14); 131.40 (C6 and C6a ↔ C10a); 130.51 (C8); 129.45 (C9); 127.00 (C10a ↔ C6a); 124.52 (C7); 123.42 (C10); 121.53 (C15 and C19); 111.24 (C4a); 79.09 (C2); 31.60 (C3); 26.77 (2 -CH3); 15.99 (C4). IR (ATR, v in cm-1): 3350 (N-H); 1703 (C=O; N-C=O); 1597 (C=N); 1558, 1511, 1493 and 1394 (C=CAr); 1230 (C-O); 918-693 (C-HAr). HRMS (ES+) calculated for C21H19N3O3 [m+H]+: 362.1504. Found: 362.1486.
Data from βHDZ (4): (E)-2,2-dimethyl-6-(2-(phthalazin-1-yl)hydrazone)-3,4-dihydro-2H-benzo[h]chromen-5(6H)-one
The reaction mixture remained at room temperature for 78 h [13]. Orange crystalline solid (figure 2), yield of 22.4 % and m.p.: 237.16-239.00 °C. 1H NMR (400 MHz, CDCl3, δ in ppm): 9.30 (1H, s, H20); 8.56 (2H, dd, J = 7.6; 3.8 Hz, H7 and H15); 7.95 (4H, m, H10, H16, H17 and H18); 7.55 (1H,m, H8); 7.45 (1H, m, 1H, H9); 2.70 (2H, t, J = 6.7 Hz, H4); 1.92 (2H, t, J = 6.7 Hz, H3); 1.49 (6H, s, 2 -CH3). 13C NMR (100 MHz, CDCl3 , δ in ppm): 181.42 (C5); 161.87 (C1a); 148.40 (C20); 132.69 (C17); 132.47 (C6 and C13); 132.40 (C16); 132.29 (C6a); 129.92 (C8); 128.17 (d9 ↔ C14); 127.74 (C9); 126.84 (C18); 125.75 (C10a); 123.51 (C7); 122.99 (C10); 122.35 (C15); 118.78 (C14 ↔ C19); 111.15 (C4a); 78.21 (C2); 31.78 (C3); 26.80 (2 -CH3); 16.19 (C4). IR (ATR, v in cm-1): 1612 (C=O; C=N; -C=N-N=C-); 1588 (C=N; -C=N-N=C-); 1501, 1437 and 1395 (C=CAr); 1225 and 862 (C-O); 900-691 (C-HAr). HRMS (ES+) calculated for C23H20N4O2 [M+H]+: 385.1664. Found: 385.1686.
Determination of the mean inhibitory concentration (IC50) of naphthhydrazones on the promastigotic forms of Leishmania amazonensis, Leishmania major and Leishmania infantum
The promastigote forms of Leishmania were kept cryopreserved in liquid nitrogen and in Schneider's0 medium (Sigma, Chemical - USA) supplemented with Fetal Bovine Serum (FBS), 100 IU-mL-1 penicillin-streptomycin (Sigma) and glycerol as cryopreservative. To perform the assays, the parasites were thawed and kept in the same medium, without cryopreserver, at 26 ± 1 °C in a biological oxygen demand oven (Eletrolab EL202, São Paulo, Brazil). The promastigotic forms of Leishmania amazonensis (IFLA/BR/67/PH8), Leishmania major (MHOM/IL/80/Friendli) and Leishmania infantum (MHOM/5745) in stationary growth phase were washed in 0.9 % sterile saline solution, counted in Neubauer chamber and the volume adjusted to the desired concentration. The substances αACIL (1), αHDZ (2), βACIL (3) and βHDZ (4) were added to the microplate wells for cell culture, in triplicate, and serial dilutions were performed, reaching twelve concentration ranges (0.0097 to 20 μg-mL-1). Soon after, the parasites were sown in the amount of1x106 leishmanias/100 μL of supplemented medium. The plate was then incubated in an oven at a temperature of 26 °C for 48h and, 6h remaining for the end of this period, 20 μL of 1x10-3 mol-L-1 resazurin was added, and the plate was incubated again. After the incubation period, the reading was performed on a 550 nm wavelength absorption plate reader (Biosystems model ELx800, Curitiba, PR, Brazil). For positive control, amphotericin B was used at a concentration of 2 μg-mL-1, diluted in a supplemented Schneider's medium. The negative control was equivalent to Schneider's medium containing 1x106 promastigotes per well and, in this case, the viability was 100 % for the parasite. The reading of white, for each concentration and for the controls was necessary to disregard the absorbance resulting from the medium itself with interference or not of the substances studied. From these absorbances the concentration able to inhibit the growth in 50 % of the parasites was calculated (IC50) [14, 15].
Determination of the mean cytotoxic concentration (CC50) of synthetic naphthohydrazone on RAW macrophages
The assessment of macrophage cytotoxicity was performed using the MTT assay (3-(4.5-dimethyl-2-thiazolyl)-2.5-diphenyl-2H-tetrazolium bromide). In the plates of 96 wells, 2x105 macrophages of the RAW 264.7 strain were incubated per well in 100 μL of RPMI 1640 medium (supplemented with 10 % SFB, 10 000 IU penicillin and 1000 IU streptomycin) in an oven at 37 °C and 5 % CO2 for 4 h for cell adhesion. The supernatant was then withdrawn to remove the non-adhered cells. The DMSO solubilized naphthahydrazonic derivatives were diluted in RPMI supplemented medium, added to the plate containing the macrophages in serial concentrations reaching twelve ranges of final concentrations, starting from 100 μg-mL-1, and incubated at 37 °C and 5% of CO2 for 48 h. After this period, the cytotoxicity was evaluated by adding 10 O MTT at a concentration of S mg-mL-1, diluted in 100 μL of RPMI medium, and the plate was incubated again for 4 h at 37 °C and S O of CO2. At the end of this period, the supernatant was discarded and the formazan crystals were dissolved by adding 100 μL DMSO. Finally, the absorbance (330 nm) was measured using a Biotek plate reader (ELx800) [16]. The mean cytotoxic concentration CC50 (μM) was determined from the linear portion of the curve, calculating the concentration of the compound that reduced the absorbance in treated macrophages by S0 O compared to negative control cells. The selectivity index (SI) was calculated by the ratio between CC50 and IC50 [17].
Statistical analysis
All the biological trials were performed in three independent experiments. The mean inhibitory concentration (IC50) and the mean cytotoxic concentration (CC50) with 9S O confidence limit were calculated using probit regression. Analysis of variance ANOVA followed by Bonferroni's test was performed taking p < 0.05 as the maximum level of statistical significance.
RESULTS AND DISCUSSION
Synthesis of the naphthohydrazones αACIL (1), αHDZ (2), βACIL (3) and βHDZ (4)
The hybrid compounds designed from α- and β -lapachones, with the isonicotinoylhydrazone and phthalazinylhydrazone nuclei were called αACIL (1), αHDZ (2), βACIL (3) and βHDZ (4) (figure 1). The prefixes α- and β- refer to lapachol derivatives, α- and β -lapachones, and ACIL and HDZ refer to the isonicotinoylhydrazone and phthalazinylhydrazone nuclei, respectively. The hybrids were synthesized by condensation reaction between α- and β -lapachones with isoniazid and hydralazine through acid catalysis, as described by Guimarães et al. (2020) [13]. The reaction yields varied between 10 O and 66 and the compounds had their molecular structures established by NMR, IV and MS techniques (figure 2).
Antileishmanial activity of naphthohydrazones
The performance of in vitro tests of antileishmanial activity only in promastigote forms has been used as a screening test [16-20]. The in vitro evaluation of the antileishmanial activity of naphthohydrazones αACIL (1), αHDZ (2), βACIL (3) and βHDZ (4) has shown that these four hybrids were effective against promastigotic forms of L. amazonesis, L. infantum and L. major, this action being dependent of the concentration of the substance in test and the species of the parasite. When comparing the results obtained with the antileishmanial capacity of the precursor naphthoquinones α- and β-lapachones) it was observed that the majority of the hybrids were more active.
Among the hybrids of a-lapachone both compounds, αACIL (1) and αHDZ (2), showed high antileishmanial activity in at least one of the strains tested in the experimental time of 48 h. However, αHDZ (2) was more potent against L. amazonesis and L. major, with IC50 of 0.156 and 0.572 μM, respectively (table 1). Naphthohydrazone 2 showed higher activity than its precursor, α-lapachone (IC50 16.08 μM in 72 h of experiment) [9] for L. amazonesis, exhibiting 103 times higher activity in a shorter experimental time (48 h). For L. infantum, αACIL (1) and αHDZ (2) presented activity 24 and 4 times higher, respectively, than α-lapachone (IC50 13.88 μM in 72 h of experiment) [9], in an experimental time of 48 h.
The most active compounds against all the promastigotic forms of leishmanias tested were those that have the framework of phenanthrene, as occurs in β-lapachone and its hybrids β ACIL (3) and βHDZ (4). While 3 had the lowest IC50 (0.318 μM) against L. infantum, compound 4 was the most potent against L. amazonesis, with a IC50 of 0.023 μM. Both hybrids, 3 and 4, were more active than the precursor compound, β -lapachone, on all tested strains of leishmanias. βACIL (3) was 65 times more active than β -lapachone (IC50 2.90 μM in 72 h of experiment) [9] for L. amazonensis; whereas, βHDZ (4) was 126 times more active than the precursor orthonaphthoquinone in L. amazonensis. Compared to L. infan-tum, hybrids 3 and 4 were 2 times more active than β-lapachone (table 1).
Table 1. In vitro antileishmanial activity in promastigotic forms of L. Amazonensis, L. major and L. infantum of synthetic naphthohydrazones, in 48 h of experimental time.

a IC50: concentration of the compound that causes 50 % mortality.
b CC50: concentration of the compound that causes 50 % of macrophage mortality.
c SI - CC50/IC50 when SI >1 means higher toxicity to the parasite.
d IC50 (μM) in 72 h (Guimarães et ai, 2013) [9].
e Tanga 2013 [21].
f Non-existent Result.
g Dias et ai., 2020 [22].
When comparing the IC50 of the synthesized naphthohydrazones with the amphotericin B, it can be affirmed that after exposure to L. amazonensis and L. major, the majority of the naphthohydrazones presented activity superior to the positive control. Highlight to β HDZ (4) over L. amazonensis and β ACIL (3) compared to L. major, both with action 15 times greater than amphotericin B. Regarding L. infantum, the compounds 3 and 4 presented activity similar to the standard drug (table 1).
Leishmaniasis are intracellular macrophage parasites in vertebrate hosts; therefore, assessing toxicity to these cells is essential when planning a drug for the treatment of visceral or tegumentary leishmaniasis [23]. The factor used to measure this safety was the selectivity index (SI), which is calculated by the ratio between the Cytotoxic Concentration for 50 % of macrophages (CC50) and IC50. The molecules that present SI > 1 and SI > 20 are classified, respectively, as good and high safety, and those of high safety are qualified for further studies in infected macrophages and in vivo models [24, 25].
The tested naphthohydrazones showed good safety for use as antileishmanial, highlighting the hybrid βACIL (3), 52 times more selective for L. amazonensis and L. major than for the host cell. The compounds that have the HDZ nucleus, αHDZ (2) and βHDZ (4), also showed high safety for mammalian cells with SI of 42,956 and 24,179, respectively, when used on promastigotes of L. amazonensis. These results qualify the naphthohydrazones αHDZ (2), βACIL (3) and βHDZ (4) for the trials on infected macrophages and in vivo models. Among the compounds tested, αACIL (1) and βHDZ (4) were the most selective for L. infantum in relation to macrophages, presenting SI of 8.821 and 8.043, respectively.
Although the mechanism of action of naphthohydrazones on leishmanias is not known, the results of this work show that the compounds promoted damage in the morphological structure of these parasites (figure 3). Among these changes, the variation in parasite size, scourge and shape stand out. According to Rodrigues et al. (2014) [26] and Gadelha et al. (2013) [27], such changes may be caused by the destabilization of the tubulin-dependent cytoskeleton, since both the parasite's body shape and the scourge's integrity are highly dependent on the stability of the microtubules.

Figure 3 Leishmania amazonensis treated with naphthohydrazones in the concentration corresponding to its IC50. (A) without treatment; (B) αHDZ (2); (C) βHDZ (4); (D) αACIL (1); (E) βACIL (3). 100x magnification. Arrows represent promastigotes with altered morphology.
Although naphthohydrazones have relatively complex molecular structures, they were prepared using accessible compounds, the lapachol, a natural product from a renewable source; and two low-cost drugs, with known toxicity and pharmacokinetics, the isoniazid and hydralazine. In addition, classic reactions in mild conditions were used, and accessible synthetic routes are desired by the pharmaceutical industry, especially when it comes to drugs for the treatment of neglected diseases, such as leishmaniasis. The promising antileishmania activity presented by naphthohydrazones, in association with the synthetic advantages of the route used, make the compounds αHDZ (2), βACIL (3) and βHDZ (4) suitable for the later stages of drug development.

